



Framing the Imprint: Omicron and Original Antigenic Sin
Imprinting has been blamed as a key factor for the lackluster responses to the Omicron spike protein. In fact, the fault may be with Omicron itself (and we might be able to do things about that).

tl;dr- Some new recent work shows that Omicron’s spike protein mutated to engage with a receptor (Siglec-9) that suppresses immune responses, and also contains a mutation that reduces its production from cells, lowering the dose of spike protein we are exposed to and making it harder to make responses against it.
A single amino acid change to stop engagement with Siglec-9 has been shown to boost antibody levels by about 10-fold in animals.
Additional recent work suggests that the recall of memory produced by our first exposure to the spike protein, the ancestral one, is responsible for the production of extremely potent, broadly protective antibodies that cover many variants, and even other SARS-like viruses, arguing that original antigenic sin is probably not our enemy against SARS-CoV-2.
The Omicron spike protein probably needs a few (very small) tweaks for our immune system’s true potential to be unleashed.
The Omicron variant fundamentally changed the pandemic once it emerged. As people1 wrung their hands about whether the Delta variant meant people should get a third dose of vaccine, Omicron rapidly began to take over and spread even among populations with high levels of immunity.
The severity of the variant was, thankfully, blunted substantially by the accumulated immunity in the population, but it spread very rapidly2. The debate about third doses was basically settled: getting a handle on Omicron’s spread would absolutely require more immunity than we had at that moment and the only safe way to get more immunity would be through vaccination.
Unfortunately, in contrast to other variants that were readily quelled by vaccine-elicited immunity, Omicron was far less accommodating. Evidence for the benefit of third doses accrued but case counts remained substantial, and even though hospitalization and death rates dropped precipitously, the absolute numbers were in many cases the highest they had been in the entire pandemic.
Eventually, Omicron’s spike protein was added to the vaccine (either in the form of BA.1 or BA.4/5), in the hopes that this would turn the tide back into our favor. Evidence was consistent in that adding the Omicron spike protein improved antibody responses against Omicron subvariants, but the extent was less substantial than was hoped, and Omicron continued to spread with relative facility (though the bivalent vaccines consistently proved helpful to those who took them).
So, has our immune system failed us or is there perhaps something more complicated going on?
Here, I argue that we were too hasty to set the blame on Original Antigenic Sin (imprinting), and substantial evidence has come to light to suggest that the Omicron spike protein itself shares a good portion of the blame (and that might have some relatively easy fixes).
First, let's delve into the concept of imprinting, which requires a bit of a journey.
Immunological Imprinting: A Rough Guide
The explanation some reached for immediately is immunological imprinting. You might also know it by the terms original antigenic sin, antigenic seniority, backboosting, negative interference, primary addiction, or persistent antibody orientation (these aren’t exactly the same thing but for the purposes of this discussion can be treated as being synonyms. It should also be noted that the terms are not consistently applied throughout the literature so if you find them confusing, you are not alone; for details on the distinctions see this footnote3). Here’s the basic idea:
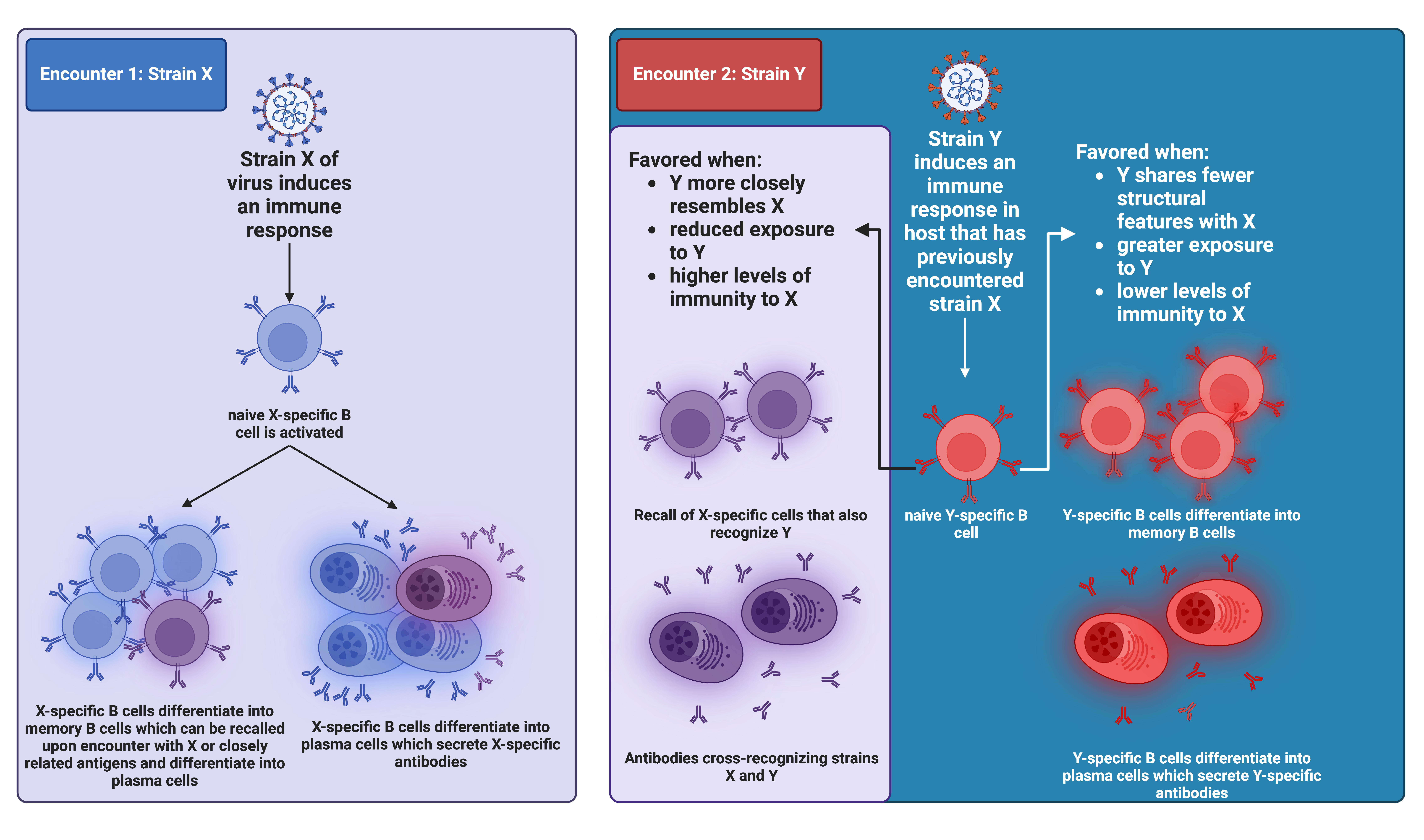
You encounter strain X of some virus (the choice of virus here is arbitrary, it can be any antigen).
You raise an immune response against it which includes antibodies against X made by plasma cells and X-specific memory B cells.
Some time passes and you now encounter strain Y of this virus. Strain Y and strain X share some structural features4 so now the immune system faces a dilemma (if you’ll pardon the anthropomorphization): does it try to recruit Y-specific antibodies and B cells from the pool of naive cells or select the memory B cells I already made against X that are also specific against Y?
Recycling the cells made in my response to strain X instead of recruiting new cells specific to strain Y is known as imprinting (or any of the other terms I mentioned before, depending on the consequence). The more similar Y is to X, the more likely I am to try to find the X+Y-specific cells than to recruit newly minted Y-specific ones.
You might reasonably wonder, why would I care where my antibodies are coming from (i.e. memory cells vs. naive cells) so long as the antibodies I make work? This is the exact right question, and I will tell you that when it comes to SARS-CoV-2, I could not care less about the source of my protective antibodies so long as they are protective. However, whether or not imprinting is good or bad depends on the context (and who you ask)5.
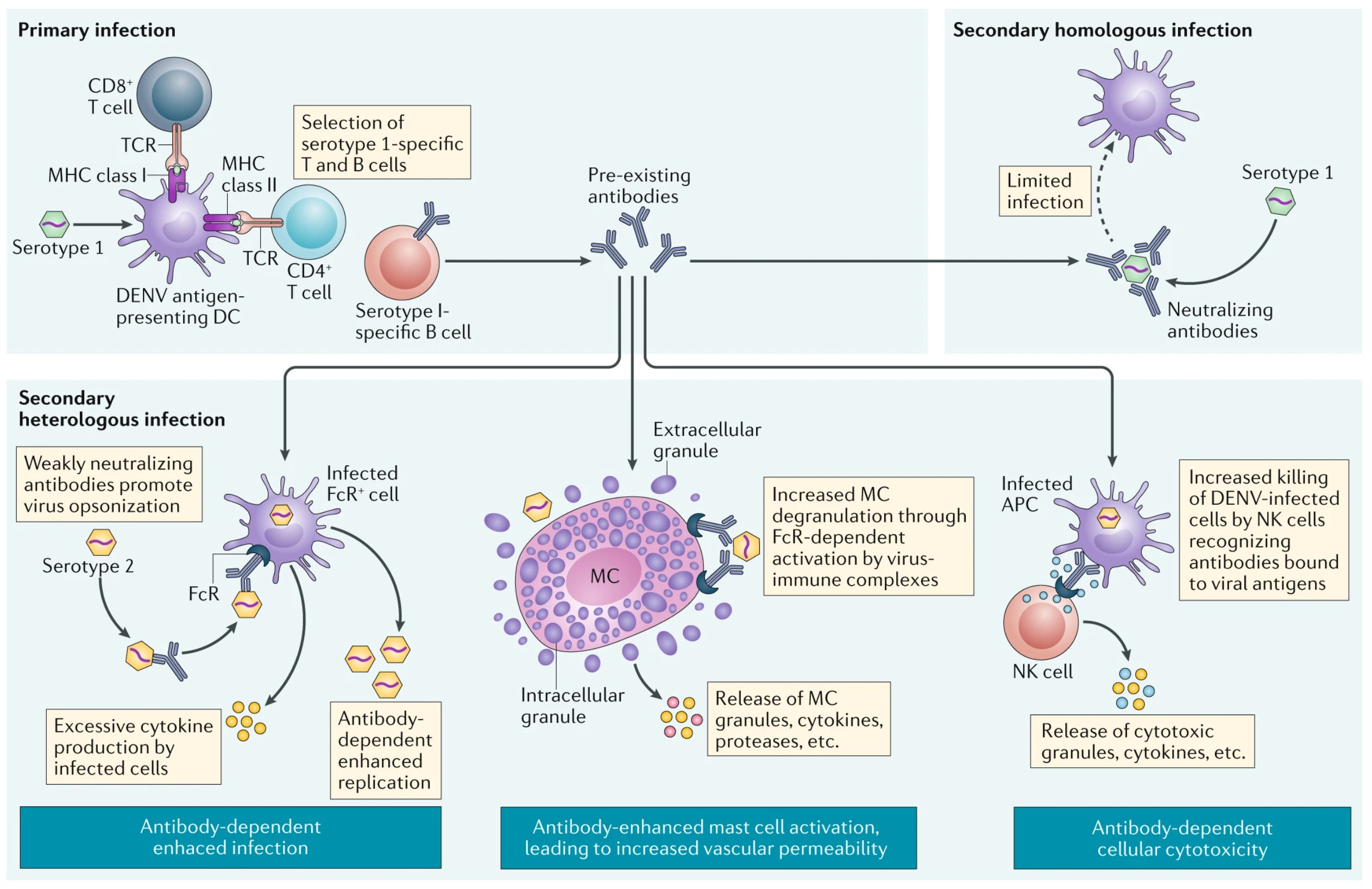
Imprinting can be clearly bad in the case of dengue infections (you may know dengue by the name “breakbone fever”), because dengue viruses can take advantage of antibody dependent enhancement (ADE). Antibodies can instruct the cells of the immune system to respond in particular ways through proteins on the surface of those cells called Fc receptors. In some cases, if the antibodies fail to neutralize dengue virus particles, they can enable increased uptake of dengue into those immune cells and also promote signaling that enhances dengue’s replication within those cells. This can cause a devastating condition called dengue shock syndrome where the massive inflammatory response that results causes leakage of fluid through the capillaries of the body and dangerous drops in blood pressure. The biggest risk factor for severe dengue and dengue shock syndrome is a prior infection with a different strain of dengue. Dengue has four strains (officially), and antibodies against one of the strains generally don’t work that well against antibodies of the other strains. If your antibody levels fall within a specific range where they bind to dengue viral particles but do not effectively neutralize them (either because they don’t bind to the right parts of the virus or because there aren’t enough antibodies), ADE will occur (something that is supported by dengue infections where the new virus falls in a goldilocks zone of structural similarity relative to the ones previously encountered). Thus in this instance, imprinting caused by infection with one strain of dengue can make infection with a second strain of dengue far more dangerous. For this reason, dengue vaccines generally have to be given only to those with prior immunity to dengue (as this keeps antibody levels above those that carry a risk for ADE) and need to give comparable levels of immunity to all four strains of dengue at the same time (something that has consistently proved to be a challenge). Fortunately, even though ADE can be shown in vitro for basically any virus given the right ratio of particles to antibody, an effect in humans where first infections make the second more dangerous is fairly unique to dengue, and hasn’t been observed with COVID-19 despite looking extensively.
While imprinting can clearly endanger people in the case of something like ADE, another concern with imprinting is negative interference (some people mean this when they say “original antigenic sin”6). In essence, the presence of that prior immunological memory against strain X (per the example above) reduces the response to strain Y. This potentially means that someone who has never seen strain X and instead just sees strain Y is better protected against another encounter with strain Y than someone who sees strain Y after seeing strain X. It does not, however, mean that someone who has never seen Y before is more protected than someone who has seen X and Y. The person who has never seen X or Y before and spontaneously encounters Y has no immunological memory to rely on to control Y and protect them; the person who has seen X will (in principle) have at least some memory that can help control the infection. In short, negative interference on its own won’t be a good reason to not get vaccinated: it’s the choice between some additional protection or none at all7.
There is one major fallacy I have seen repeated many times over throughout the pandemic with regard to imprinting and it merits attention:

Suppose the immune system encounters strain X multiple times and then encounters strain Y. We observe that the antibody response to strain Y is less than that of strain X- is imprinting to blame for the lower response? While this doesn’t exclude a role for imprinting in that lower response to Y than X, this type of experiment cannot tell you that imprinting is disrupting the response to Y. First of all, the comparison is not parallel: assuming X and Y are equally good antigens for the immune system (i.e., they give similar antibody responses in organisms who have seen neither before), we would always expect antibodies against X to be higher than those against Y because we are comparing the effects of a single exposure to Y to multiple exposures to X. By virtue of this factor alone, no conclusions about imprinting can be drawn simply by comparing antibody responses to X and Y in a group that has been exposed to both.
Immune Mechanisms Underlying Imprinting
At the basic level, for imprinting to occur, there have to be conserved structural features across the antigens in question- if the memory cells can’t recognize anything on the new antigen, no imprinting can occur. Thus, to the extent that imprinting poses a problem, (in a vacuum, i.e. not considering things like antibody-dependent enhancement) it is more-or-less a self-correcting one: the more the new antigen differs from what the immune system has previously encountered, the easier it will be to recruit naive B cells to make a response specific to that antigen. Experiments in mice show that when encountering the same antigen as one previously seen, essentially all of the antibody response comes from B cells activated during the initial encounter (if it ain’t broke, right?).

Imprinting is fundamentally based on the ability of memory B cells to outcompete naive B cells. Since memory B cells have already previously been activated, meaning that the immune system has judged them to be safe enough to survive selection, their activation requirements are much less stringent than those of naive B cells that have never been activated before. This means that the natural tendency of the immune system is to recruit memory B cells over naive B cells. That does not however mean it is impossible to activate naive B cells- they just need more help to be recruited into immune responses. Thus conditions which are more stimulating to B cells have a higher chance to activate naive B cells can help reduce the effect of imprinting, such as the use of adjuvants.

The second major mechanism implicated in imprinting is what is known as antibody feedback. Firstly, it’s critical to remember that the cells in our body have receptors for antibodies known as Fc receptors (because they recognize the Fc region of the antibody- the stalk part), and these receptors make the cells that have them do things. B cells all have an antibody receptor called CD32B or FcγRIIB (γ because it recognizes IgG antibodies), which is an inhibitory receptor (the only inhibitory type I Fcγ receptor) and no other type I8 Fcγ receptors9: upon binding antibodies (or, more properly, immune complexes), this receptor sends an inhibitory signal to naive B cells that can suppress its differentiation into a memory B cell or plasma cell, and potentially even make the B cell die by apoptosis if the signaling is strong enough. B cells from patients with autoimmune disease seem to lose expression of CD32B. It should be noted however that antibodies can also directly contribute to the diversification of targets by the immune system through antibody feedback as well. When antibodies bind, they necessarily obscure the epitope that they bind (literally, it is physically covered by the antibody and so another antibody cannot bind it or epitopes that overlap with it; this is known as epitope masking), which means that for B cells to bind to the antigen, they must target new epitopes that are not obscured. This allows the immune system to focus on what are known as “subdominant epitopes” to contrast with the epitopes that the immune system tends to focus on normally (immunodominant epitopes)10. This means that you can still see diversification of the immune response to target new parts of the antigen even if you repeat immunization with the same exact antigen. In fact this is exactly what happens with the vaccines: the S2 part of the spike protein, which lies closer to the membrane, is better conserved across coronaviruses, and so with the first dose of vaccine, most antibodies are directed against it as a recall response occurs. With the second dose, these epitopes are now masked, allowing the immune response to re-orient and focus on S1, where the most potent neutralizing epitopes lie:

Perhaps most important factor here is the role of antigen dose. Imprinting in general focuses on B cells, but this does not mean B cells and their descendants are the only actors important in the immune response. For example, after a viral infection (or vaccination with an mRNA vaccine), CD8 T cells are induced which can kill infected (or transfected) cells. If this occurs before the immune system has a chance to raise a generation of naive B cells against the antigen, naturally, the antibody response that results will be smaller. Antibodies can also recruit other cells to kill infected cells, such as natural killer cells, monocytes, and macrophages. Together these all have the effect of reducing the amount of available antigen. In the same vein, when antibodies recognize antigen and form immune complexes, these are taken up by cells that bear receptors for antibodies, also reducing the dose (antigen trapping). Using higher doses of antigen can help ensure that naive B cells can encounter at least some, so this can also help with alleviating imprinting (but this is at times counterproductive, such as if the goal is a large immunization campaign- ideally you want to immunize the maximum number of people which means using the least amount of antigen necessary to be protective; here adjuvants can help by allowing for dose-sparing).
I should also mention that one paper sometimes cited on the topic argues that regulatory T cells are key to this process. Regulatory T cells essentially serve as brakes for the immune system, serving to limit its tissue damage in the course of immune responses and blocking immune responses against self-antigen so that people don’t get autoimmune disease. While this explanation fits with the outcomes reported in studies on original antigenic sin insofar as there may be negative interference, I am not aware of direct, in vivo or in vitro evidence that regulatory T cells are important players in this process and available evidence does not suggest they are essential for it.
Imprinting and SARS-CoV-2
We know that the Omicron variant of SARS-CoV-2 gives less robust immune responses than older variants of the virus (well, at least if you look in blood). The issue is Omicron emerged at a time when a huge proportion of people had spike-specific memory B cells, and that proportion of people has continued to rise- how do we know then that the immune responses aren’t because of Omicron itself, rather than our immune systems’ failures to plan ahead? As it happens, we have now amassed significant evidence that is in fact IS Omicron’s fault. Here are the main factors:
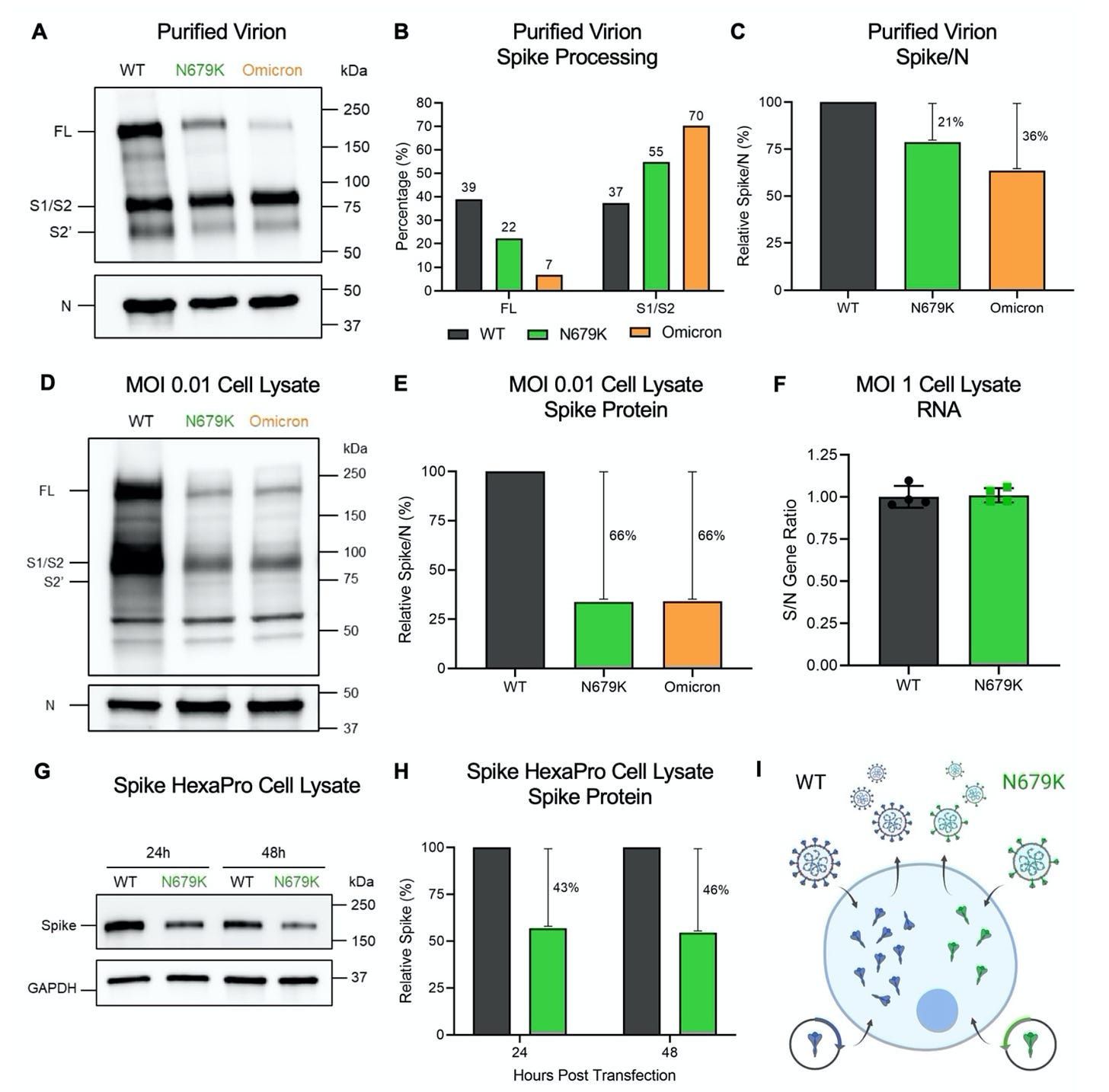
Omicron spike is less efficiently produced by cells because of a mutation: We kind of take for granted that to the immune system it’s all spike proteins, and that’s true to an extent- but it’s less true when the spike proteins actually have to be made in the cells. The N679K mutation in the spike protein results in reduced amounts of spike protein being made both inside cells and on viral particles. This makes it harder to get to the necessary dose of spike protein by either infection or mRNA vaccine, and thus makes the resulting immunity weaker.

This mutation is, by the way, present in everything since Omicron (a handful of sequences are there with Y, R, and T):

Omicron spike engages with an inhibitory receptor, Siglec-9: On some level, the immune system is just a very complicated set of “on” and “off” switches on machines that destroy cells and biological macromolecules (I mean they do other stuff too, but you don’t go to Vegas to see the fountains11). Siglecs are proteins that recognize sugars known as sialic acids, and in turn serve as “off” switches for the cells that bear the siglecs. Omicron evolved a spike mutation, F375, which allows it to now engage the protein Siglec-9, which is present on multiple cells of the immune system. This in turn suppresses the function of multiple immune cells, but in particular markedly reduces the ability to present the Omicron spike protein by phagocytes. How potent is this effect on the immune response? The team reverted F375 to S375 to prevent engagement with Siglec-9 and compared the antibody responses from versions of the spike protein that had the mutations to those that did not. Those bearing S375 (the pre-Omicron version) induced about 10-fold higher antibody responses against spike compared with those who had F375 (the exact fold-change varied by variant). Additional tweaking by the team was shown to further enhance the response.

This mutation is, by the way, present in everything since Omicron:

SARS-CoV-2 Omicron replicates much more quickly in the upper respiratory tract: There are a lot of references that discuss this so I just picked a recent one, and I hope people won’t take offense for not picking their preferred reference. As I have said before, when it comes to the immune system, kinetics is everything and everything is kinetics. When infections get faster, it takes a bigger immune response to catch up to them in time to interrupt spread. Omicron turned SARS-CoV-2 into more of an upper respiratory infection than prior variants. This is good news on the severity front as it makes pneumonia and hypoxemic respiratory failure less likely (unfortunately though, not nearly as much as we would want it to). It is very bad news on the “bringing this thing under control through herd immunity” front. Incidentally, this is why I was very annoyed when some manufacturers attempted to assign effectiveness values for their vaccines based on antibody responses using data from the original trials conducted in 2020 when the virus was a lot slower- the basic shape of the curve may still be true but the values in question are absolutely not (and hopefully this lends additional clarity into why it is so hard to pin down an absolute correlate of protection for SARS-CoV-2 even though we have extensive evidence that antibody responses are protective).



In sum, we have been very quick to wag our fingers and blame our immune system and our vaccines for not ending SARS-CoV-2 once Omicron showed up, but we have by comparison essentially ignored the role of the virus in all of this. I guess it’s because the former seem more in our control than the latter, but the two things are imbricated and we can adjust based on the behavior of this virus. I believe at this point it’s pretty clear that the standard Omicron spike protein is just not a very good immunogen and it requires a decent amount of help for it to be able to unleash the true potential of our immune system’s protection- which is something that can be done relatively easily (depending on regulator buy-in).
Having said that, there is perhaps a spot where we erred, in a relative sense. At the time that Omicron emerged, lineages derived from the delta variant had essentially swept the competition from other variants. The arrival of Omicron was shocking in large part because Omicron was not descended from the Delta variant. It came from a much earlier branch. The question then became: could this happen again? Spoiler- yes: After the reign of recombinant Omicrons like XBB variants, BA.2.86 emerged from a much older branch of the Omicron tree and then eventually evolved into JN.1 and the most modern subvariants. Still, given that possibility, regulators had a valid concern: dumping all the eggs into an Omicron BA.4/5 (or BA.1) basket could make for trouble if something really different emerged. Thus they made the choice to recommend the vaccines be bivalent: half ancestral variant, as that one had already shown the ability to induce robust immune responses, and half BA.4/5 or BA.1 (depending on where you were in the world). That was a reasonable thing to do with the evidence at the time, but it turns out that this actually really interferes with the immune response through antigenic competition. This goes back to the problem of antigen dose. Firstly, we now know that the ancestral protein is translated at much higher yield than Omicron is, so getting a proportionate dose of Omicron spike to be made in the body by an mRNA vaccine is probably impossible if you simultaneously introduce equal amounts of mRNA for both. However, the immune system doesn’t just sit idly by as your cells make spike protein to wait for you to make a high level. It is extensively primed by your prior immune memory against the spike protein and will seek out and destroy the cells that bear it as it is meant to do. You could therefore imagine a situation in which a cell takes up vaccine with mRNA for both BA.4/5 spike and the ancestral spike and kills it before you get a chance to make much BA.4/5 spike at all. Thus, it becomes much harder to generate a response against the novel BA.4/5 spike when it is paired with the ancestral spike protein. My takeaway here is that if we want to go with multiple antigens for a given vaccine where all of them will be presented together, we are best served by selecting antigens that are meaningfully different from the ones that our immune system is already extensively trained against (for example, a bivalent of KP.2 and SARS-CoV-1 spike).
Wait- Imprinting is… Good?
I would argue, at least when it comes to COVID-19, imprinting is, in fact, good. Basically since Omicron emerged, there has been a clarion call for vaccines that induce antibodies that would cover all variants of SARS-CoV-2, SARS-like viruses, coronaviruses, etc. Given that SARS-CoV-2 is the third coronavirus to emerge in the 21st century and cause public health devastation, the need for such vaccines seems… obvious. So the question becomes: how does one go about getting these cross-reactive antibodies? You’re not gonna believe this (but you should):
This work shows that antibodies from the very first exposure to ancestral spike protein acquire binding breadth to neutralize even the highly drifted XBB variants.
This work also shows that antibodies from the ancestral spike protein are adapted to recognize highly drifted variants. Consistency in results across multiple groups is always nice because it supports that the thing you’re looking at isn’t a mirage.
This work even identifies an antibody induced in someone with two doses of the Pfizer vaccine who then had a breakthrough infection that neutralizes every SARS-like coronavirus tested with binding so potent that it exceeds what was previously believed to be the theoretical limit on the binding capacity of an antibody. I can’t speak for everyone, but I would like approximately a buttload (highly technical immunology unit) of those antibodies all over my body (especially my upper respiratory tract).
Conclusions
Unless it’s about dengue or a few other infectious diseases, worrying about imprinting is not a useful exercise for anyone who’s thinking about vaccination unless they themselves are designing the vaccines. The bottom line is that updating your immunity to SARS-CoV-2, influenza, etc. will always have a benefit regardless of immune history. How that benefit compares to the risks of the particular vaccine and how it varies as a function of your unique immune history is another matter, but in general, I haven’t seen compelling evidence that the net result is negative for any particular age-sex stratum when it comes to COVID-19 vaccines. We should stop thinking of imprinting as a boogeyman to be feared or an obstacle to work past, and instead consider how we can work with our immune history to maximize our protection.
I am among “people” here. I spent a few weeks poring over the data regarding whether a third dose of vaccine was the right move against Delta. I had basically no doubt that it could help but I did doubt that it would help more than first doses in those who had none and the horrible inequities of the situation were salient for me (i.e. in the US people were thinking about third doses as many lower and middle income nations were struggling to even get first doses). It eventually became clear that the contracts from the US’s purchases did not allow a transfer of doses to LMICs, which meant that the inequities of the situation, while awful, were no longer material considerations (the shots would go into American arms or they would be trashed). Further, while I felt we could and should try to get the vaccine to those who hadn’t had any doses, at that point in the pandemic it was widely available to adults. Barring access issues, those who wanted the vaccine could get it. I eventually concluded that third doses were the right move and prepared to write up the evidence synthesis on my blog… and then news of the Omicron variant emerged and I ended up not writing that post because it was no longer relevant.
Many have at points claimed that Omicron is a fundamentally milder infection than other variants of SARS-CoV-2 and this is the major reason that COVID-19 has become less deadly. Depending on how the point is specifically phrased, this claim ranges from being misleading to untrue. When Omicron attacked populations where there weren’t high levels of immunity, the outcomes were catastrophic. Further analyses suggested that Omicron was less virulent than Delta, but about the same as the ancestral variant of SARS-CoV-2 (the virus that started this whole pandemic). Immunity is the much more important factor in the attenuation of SARS-CoV-2’s severity.
Even though the term is probably most famous and commonly said to have been the first description, “original antigenic sin” actually came after “persistent antibody orientation.” way back in 1956 by Jensen and colleagues. I don’t think this term has ever really been used by anyone since, but basically, the group noted that if they first adsorbed antibodies against a flu virus that the hosts had encountered earlier, most of the reactivity to the new flu virus went away, suggesting that responses to the new virus were driven by crossreactive antibodies.
Francis followed soon after with the term “original antigenic sin” or OAS in 1960, then meaning a case in which there was a greater antibody response to the priming immunogen (i.e., strain X in the example) than the boosting immunogen (i.e., strain Y in the example). Unfortunately, this term has been used inconsistently throughout the literature to the point that it’s hard to say what it even means. For example:
Our current findings demonstrate that adjuvants relieve original antigenic sin by inducing cross-reactive memory B cells.
While the concept was coined before there was a molecular understanding of the nature of the immune response, this paper was not- we know today that cross-reactivity underpins the phenomenon of imprinting, OAS, etc. and thus to claim that cross-reactive memory B cells alleviate the phenomenon makes for a very confusing sentence (to the best of my ability to tell, the meaning of the sentence in context was something like “recruiting cross-reactive B cells allows for effective antibody responses to boosting immunogens”).
The recall of antibodies targeting older variants of a particular antigen is also known as back-boosting (because it is a boost to target variants that were encountered backwards in time). Backboosting suggests that the responses arises from old immunological memory (but does not prove it- to do that you have to actually sequence the BCRs from the immune responses and see how mutated they are). Backboosting can also complicate interpretations of immunological data. For example, in what my friend Dylan Morris has referred to as “perversity,” the pre-existing immunological memory serves to suppress effective responses against a new immunogen. Intuitively you might think that one way this could be seen is if the antibody response to the new immunogen induces much higher antibody titers against the old immunogen than the new one- but even this does not prove such a thing. Why? During the course of immune responses, antibodies mutate to bind more tightly and cover more variants of the immunogen, and these two properties can coincide. Thus, it is possible that upon boosting with a new immunogen, the broadened cross-reactive antibodies that result may also have heightened affinity for the priming immunogen compared to their prior state.
Antigenic seniority is perhaps easiest to understand with a visual aid:

In essence, under the idea of antigenic seniority, there is a persistent recall of the very first exposure, but there is also consistent addition to the pool of responsive cells and antibodies by subsequent exposures to different but related antigens. Under primary addiction, or OAS, the response is always dominated by cells and antibodies produced from the first exposure, which continue to evolve but essentially overwhelm the contribution of any new, naive cells.
Usually the degree of antigen similarity (or dissimilarity) is not determined purely by structure but by a metric known as antigenic distance. This can become involved but in essence one can construct a map using antisera from donors and a pool of antigens and measure their reactivity with some antigen (the one the antibodies respond to the strongest), and the other antigens positioned away from that reference antigen in proportion to the amount that antibody recognition of that antigen drops. This is known as antigenic cartography. The antigenic distance between two identical antigens would be 0. This describes in greater detail how one would measure antigenic distance:

In this dataset, animals are immunized with a given antigen and their serum is measured for the reactivity to that antigen as well as others. To use the example on this map, if we begin with the LE antigen, we know that BN must be 5 units away, JO must be 6 units away, BE must be more than 5 units away, and HK must also be more than 5 units away. From there we can consider the antigen BN and see that LE is 1 unit away, JO is 6 units away, BE is >5 units away, and HK is 3 units away. We iteratively repeat this for every antigen and every serum until we find the points of intersection which will position the antigens on the map. The farther away two antigens are on the map, the more poorly antibodies against one of those antigens would be expected to recognize (or neutralize, depending on what type of antibody titers are being measured) the other one.
The 1918 influenza pandemic is worth commenting on here for a moment. The 1918 influenza pandemic is noteworthy for many reasons, but one is the unusual epidemiological quirk of its W-shaped mortality curve, wherein the extremes of age, but also those aged 20-40 experienced a markedly higher death rate than the ages in between. The explanation generally offered to explain this unusual result is that in the ~1890 pandemic, the H3N8 strain of influenza was predominant, resulting in a memory response that did not offer good cross-protection against the 1918 flu pandemic, meaning immunological imprinting accounts for the increased mortality. This strain was then replaced by an H1N8 strain (which also preceded it), which was followed by the mother of all pandemics- H1N1 in 1918:

This reconstruction reproduces the general shape of the mortality curve seen in 1918. Some have taken this result to mean that exposure to influenza viruses of a different subtype early in life fundamentally compromises protection against antigenically distant viruses, but there’s a problem with that interpretation: the mortality curve can just as easily be explained by a protective effect from exposure to H1 viruses before the 1918 pandemic rather than a harmful effect of H3N8 immunity. The only conclusion that can be drawn here is that exposure to an H1 virus is more protective against H1N1 than exposure to H3N8. To evaluate this, we would need a group with matched characteristics that was exposed to neither H3N8 nor H1N8 to compare how they fare against 1918 H1N1 influenza vs. those who were exposed to either.
Original antigenic sin sounds fundamentally negative, but in Thomas Francis Jr’s paper describing it, its likely he didn’t intend it that way. Francis observed that people’s immune systems tended to recall antibodies against flu strains they had encountered earlier- the sin was that people’s immune systems could not be exposed to multiple strains at the same time so that they would have a broad foundation for responses against future strains. He did not propose that it was a fundamentally harmful feature of the immune response, as some have taken it to be.
I worry that this comment might be taken out of context so I wish to clarify that considerations of whether a given vaccine dose is worthwhile should also take into account the risks of vaccination as well. It is always about the balance of risks vs. benefits; however, negative interference should not be treated as synonymous with “no potential benefits.”
B cells do have a type II Fcγ receptor- CD23, the low affinity IgE receptor. Despite its name, the low-affinity IgE receptor has a lot of functions unrelated to IgE. Some papers also report expression of DC-SIGN, the other type II Fcγ receptor, but this is not classically considered to be expressed on B cells (it literally has “dendritic cell-specific” in its name, which I recognize does not prove it is only on dendritic cells, but regardless, it’s not generally considered one of the B cell Fc receptors).
In some people, CD32C or FcγIIC occurs on the surface of B cells. This protein is not normally expressed because it has been turned into a pseudogene in most people because of a premature stop codon, but in 7-15% of the population, there is a single nucleotide polymorphism that converts this stop codon into a TAG that specifies the amino acid glutamine. whereas CD32B is an inhibitory Fc receptor, CD32C is an activating one. The presence of this protein promotes B cell activation and differentiation into antibody-secreting cells, leading to more robust antibody responses. However, this comes at the hefty price of a heightened risk of multiple autoimmune diseases, as CD32B is a key control mechanism against this, and thus anything that interferes with its function therein will promote the risk of autoimmune disease.
This is a very important consideration in looking at influenza literature, and in particular responses against the hemagglutinin protein, the protein influenza viruses use to enter cells. Hemagglutinin broadly consists of a head that recognizes sialic acid glycans and a stalk which contains the fusion machinery of the virus. The head is immunodominant to the stalk- antibody responses will tend to concentrate on the head. This is probably because the head is more readily accessed physically. Antibody responses to the hemagglutinin protein are commonly measured with a hemagglutination inhibition assay (HAI), which measures the ability of hemagglutinins to make red blood cells clump up by binding sialic acids on different blood cells. Antibodies against the hemagglutinin heads can interrupt this process. However, antibodies that bind the stalks of hemagglutinin generally cannot be detected by this assay because they generally do not interfere with binding to sialic acid glycans. The stalk of the virus is much better conserved than the head, which means that over successive exposures, we build up a repertoire of hemagglutinin stalk-specific memory B cells. It is at times reported that successive immunization with influenza vaccines results in declining HAI titers, which some have interpreted as proof that original antigenic sin with negative interference is occurring. However, as this assay is blind to stalk antibodies, a true harmful effect on the immune response is not necessarily a justified conclusion.
I would like to clarify for anyone who works on the functions of the parts of the immune system that do not directly relate to host defense that I think your work is fascinating and really important and the comment this is footnoting is intended to be facetious.
Many thanks Edward!
This is Shannon Dickson here on early Monday morning, June 17th. And I been impressed through out the pandemic, with your in-depth insights, such as found in your most recent 'Framing the Imprint: Omicron and Original Antigenic Sin', an amazing article!
Also, I would much appreciate the opportunity to have a relatively brief phone call with you at some point, at your own convenience and whenever you have a bit of free time, from roughly say, 15 minutes to half and hour tops ... if possible.
I'm also hoping you might be able to connect me with Dr. Paul Offit at some point for a similar brief connection by phone. I much admire you both, as you may well already know.
I'm 72 years old and my wife, Magdalena, and I are in the same age group.
And I am president of Afibbers.org, the longest running patient education and advocacy resource online dating back roughly 30 years now, with my main focus our advocacy and education in Cardiology and Electrophysiology, in helping our large group of 'Afibbers' making their best choices toward minimizing and improving Atrial Fibrillation around the U.S. and Canada.
One last thing, you might find interesting is that I contracted oral polio at 10 yrs old in the fall of 1962 from the Sabin vaccine during the initial 'Sabin on Sundays' oral polio campaign in the U.S. to inoculate as many folks from polio as possible back in 1962.
From then on, polio has been a significant part of my life. And the great care and compassion I experienced during the roughly 2 years from 1962 of dedicated physical therapy as I partially recovered over time with the wonderful doctors and PT nurses in the Houston Medical Center back in the day with large polio clinics.
One aspect is for sure, you would be very hard pressed to find any actual polio patients who would have ever become an 'antivaxxer' ... not with all the great care and compassion we all experienced back in the day that cemented our natural love for public health and science! :-).
Cheers!
Shannon Dickson