



A Primer on Antibodies
A lot of what I discuss on this blog relies on an understanding of how antibodies work. This post attempts shorten other posts by offering a summary.

This post is one of my “vegetable” posts (to contrast with the “dessert” posts). My audience seems to be variable in their level of comfort with immunology and I want to make sure that those who have had less exposure to it can still follow along with my other content, so this attempts to level the playing field some by providing background on how antibodies work. The footnotes are more technical and should be treated as optional details for those who want to know more.
Antibodies are proteins that bind antigens (literally antibody generators- treat “antigen” to mean “something that the immune system is capable of recognizing”). An immunogen is an antigen that generates an immune response and can usually be treated as a synonym for antigen1, but even though it’s more precise, people still tend to use “antigen” for everything. Immunogenicity is the ability of something to trigger an immune response. Usually, we think of antigens as being proteins because T cells only recognize protein antigens2 and T cells are usually needed to make antibodies, but when it comes to antigens that can be recognized by antibodies, they can be basically anything: sugars (polysaccharides), fats (lipids), DNA, RNA, even inorganic compounds- you can, under the right circumstances, raise antibodies to all of them.
Formally, the antibody is a secreted B cell receptor (BCR3 as distinct from the BCR that exists embedded in the membrane of the B cell); the terms immunoglobulin (sometimes abbreviated “Ig” as in IgG) and BCR describe both an antibody and the membrane-bound BCR. It is estimated that humans are capable of making a quintillion (that is 1,000,000,000,000,000,000) distinct antibodies. The part of an antigen that an antibody binds is called an epitope. The part of the antibody that binds the epitope is called a paratope (the tips of the Fab). The paratope contains hypervariable or complementarity determining regions (CDRs- so named because they determine whether or not the interaction between the epitope and paratope is “complementary.” Think how a key has to fit a keyhole4).

Classically, antibodies are described as being specific for a single epitope or antigen but in practice that’s often not totally true. Antibodies can sometimes cross-react to multiple epitopes, which can be good or bad depending on the context. For example, if there is an epitope on a microbe that varies a lot across different variants of the microbe, a high level of cross-reactivity could mean broader protection. On the other hand, extensive cross-reactivity could lead an antibody to target substances found on healthy cells, leading the immune system to attack them. In some cases, microbes can induce these by having antigens that resemble molecules in our own cells, which is known as molecular mimicry.
The amount of antibody in a sample is called antibody titer. The measurement of titers is done through titration5. Titer can also used to describe the amount of virus in a given sample. With every immune response involving antibodies (sometimes it doesn’t get that far because the innate immune system can handle it before you need to involve them), there is initially a large peak of antibodies after exposure which eventually declines to a stable level that remains much lower than the peak, as represented below.

Antibodies are made by antibody-secreting cells (ASCs, a term encompassing plasma cells and plasmablasts6) which come from B cells. Formally, B cells themselves cannot secrete antibody until they differentiate into an antibody-secreting cell, which may happen when they are stimulated by the antigen they are specific to7. Antibody-secreting cells differ dramatically in their lifetime and ability to secrete antibody. Plasmablasts are short-lived cells made early in an immune response that provide a burst of antibodies to help control the infection before either dying or differentiating into plasma cells. Plasma cells produce much more antibody than plasmablasts and can be short-lived or survive for the rest of our lives in the bone marrow (and, as it turns out, in our mucosal tissues too). The massive peak in antibody levels seen shortly after vaccinations and infections overwhelmingly comes from the plasmablasts and short-lived plasma cells. The number of long-lived plasma cells dictates the antibody titer we have in the absence of exposure to an antigen, ensuring it is doesn’t fall to 0 (so long as we have the plasma cells).
Before going further we need to understand a bit about the structure of an antibody:

In general, most of antibody function can be broken down into the role of just 2 parts: the Fab (fragment antigen binding- the tines part, if you think of an antibody like a fork) and Fc (fragment crystallizable- the stalk part that you would use to hold a fork). The Fab regions determine what the antibody binds to (containing the variable regions of both the heavy and light chain- the part that give the antibody its specificity). Many cartoons incorrectly draw antibody binding occurring between the Fabs, giving the antibody a single binding site. Here is how antibody binding of antigen actually works:
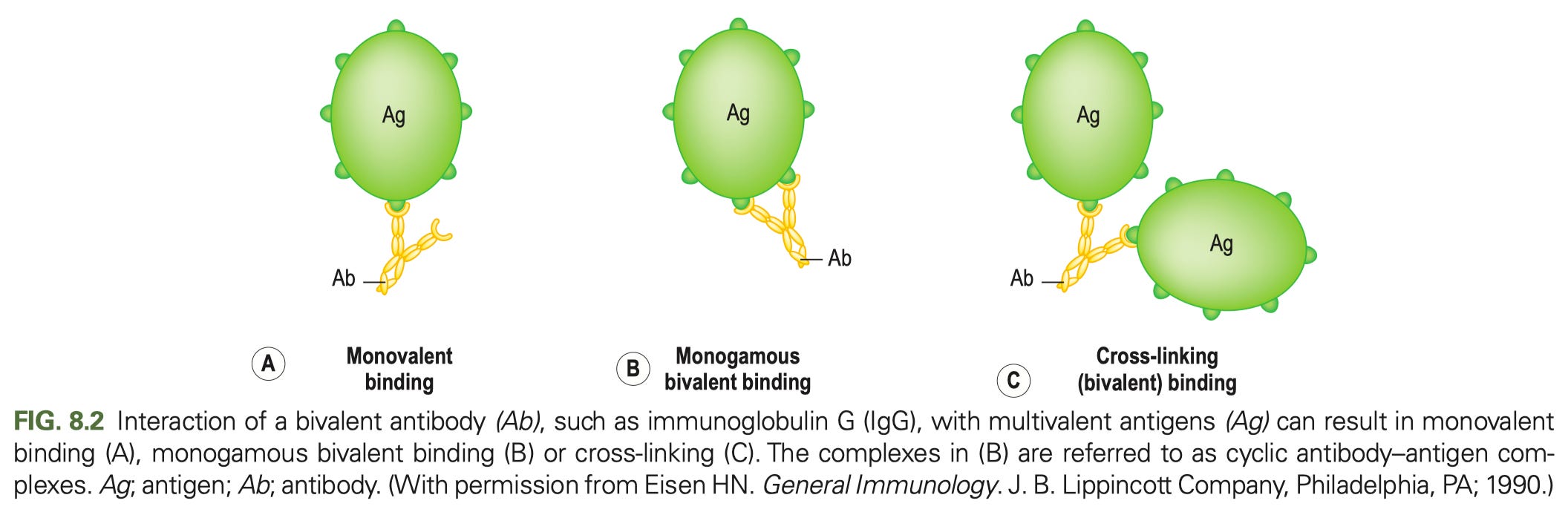
Each Fab is capable of binding an antigen, and binding should be drawn accordingly (one of my pet peeves is when cartoons draw antibody binding occurring between the Fabs at the hinge- this is not only inaccurate but it would have profound biological consequences if this were actually how it worked). The Fc region binds Fc receptors8 on immune cells triggering different types of responses from them (such as uptake of the antibody and bound antigen, production of cytokines, or killing infected cells).
We additionally group antibodies in terms of classes (also called isotypes) based on what constant region the heavy chain uses, located in the Fc region. These differ a bit by the species. In humans, IgM, IgD, IgG, IgE, and IgA are the 5 classes in humans. Of these, IgG has 4 subclasses and IgA has 2. Different classes and subclasses of antibodies have different functions (even if they have the same Fabs) because they bind different Fc receptors (or the same Fc receptor but in different ways); the differences are discussed in detail in the footnote9. In addition to the different classes of antibodies, antibodies can get adorned with different types of sugars, which in turn has consequences on their properties as well.
We have two ways to describe the binding of antibodies in the immune system. Affinity is a measure of how tightly binding between two things occurs, usually measured as a dissociation constant, with lower values meaning tighter binding. Association constants can also be used (this is the reciprocal of the dissociation constant) but in that case, higher values are indicative of tighter binding. Both of these constants reflects the balance in the sticking together of the two things (association) and coming off (dissociating). In biology, everything sticks to everything, it’s just a question of how much the laws of physics and chemistry favor sticking vs. unsticking.
Avidity is the sum of all binding interactions in a given situation. Most of the Fc receptors have low affinity for antibodies, but when the antibodies exist as immobilized immune complexes (simply lattices of antibody and antigen; more on those in a moment), the avidity of these complexes can enable tight binding, which then enables signaling (this is exactly what it sounds like- how your cells talk to each other). Ideally, an immune response from a vaccine or infection elicits high affinity antibodies which come together to give high avidity that inhibits the function of the thing they bind to10.
Immune complexes can vary in their size (depending on the concentration of antibody and antigen) and size is a key determinant of the properties of these immune complexes as the figure above shows (the bits about signaling and effector function will make more sense later as you read). The size of an immune complex is determined by how much antigen and antibody are present. If there is a large excess of antigen relative to antibody or antibody to antigen, immune complexes will be small. Larger immune complexes are more potent activators of immune cells.

As you make immune responses, antibodies undergo 2 major changes (details for the process in11). The first one is known as affinity maturation. This is where the paratope of the antibody evolves to bind more tightly to the antigen, which is achieved through mutation of the variable regions of the antibody (known as somatic hypermutation). In addition to making antibodies bind tighter, affinity maturation can also give antibodies that bind their target antigen more broadly i.e., so that they are more resistant to mutations within the epitope. The other change is called class switching. All immune responses begin with IgM antibodies, and then the B cells splice out the M constant region and replace it with G, E, A, or (very rarely) D12 constant regions depending on the cues they get from their external environment (which in turn is dictated by the type of threat the immune system faces). This dramatically changes the functions of the antibody, including how long the antibody is able to remain in the blood, what tissues it can access, and what cells it interacts with.
From here I must make things just a little complicated. Often when people talk about antibodies, they talk about neutralization. This is what it sounds like: an antibody binds and stops the toxic effects of whatever it’s bound to- a toxin, a virus, bacteria, etc. In the case of a toxin, this means the toxin never gets to interact with its target to cause the toxic effect. In the case of a microbe, it means it never gets to cause infection or spread from the site it has already infected. Neutralization is great because it is (in general) definitively protective- but this concept is more complicated than it sounds. Neutralization depends on the both binding avidity and where the antibody binds- it cannot be assumed that an antibody that binds to a target is neutralizing (in fact, antibodies exist that can bind in such a way that they can enhance the infectivity of microbes- but this doesn’t automatically mean they make disease worse). For example, antibodies that bind SARS-CoV-2 spike protein over the the part farthest from the virus membrane where it interacts with its receptor ACE2 are neutralizing (see figure 1c in the image below). However, the antibody binds to the side of the spike protein, chances are it can’t prevent this interaction, so there’s no neutralization. But, this turns out to be a bit more complex than it sounds at first blush. For example, some antibodies bind a particular epitope on the spike protein on the side that causes it to disassemble (see figure 1a below) which means that spike protein can no longer enable virus to enter cells- this is neutralization. Alternatively, some viruses (like influenza) use one protein to enter cells (hemagglutinin, HA, in the case of flu) and another to exit them (neuraminidase, NA, in the case of flu). While we usually think of targeting the entry protein only for neutralization, neutralization can be achieved by targeting the exit protein too- this is also stopping new infections, so it is neutralization (and these antibodies are definitely protective in the case of flu). You can even make antibodies that target the receptors virus use for entry as a strategy to neutralize them13. In the same vein, some viruses have separate proteins for attachment and entry by fusion viruses (paramyxoviruses like measles and pneumoviruses like RSV for example), and so targeting either protein can give neutralization.
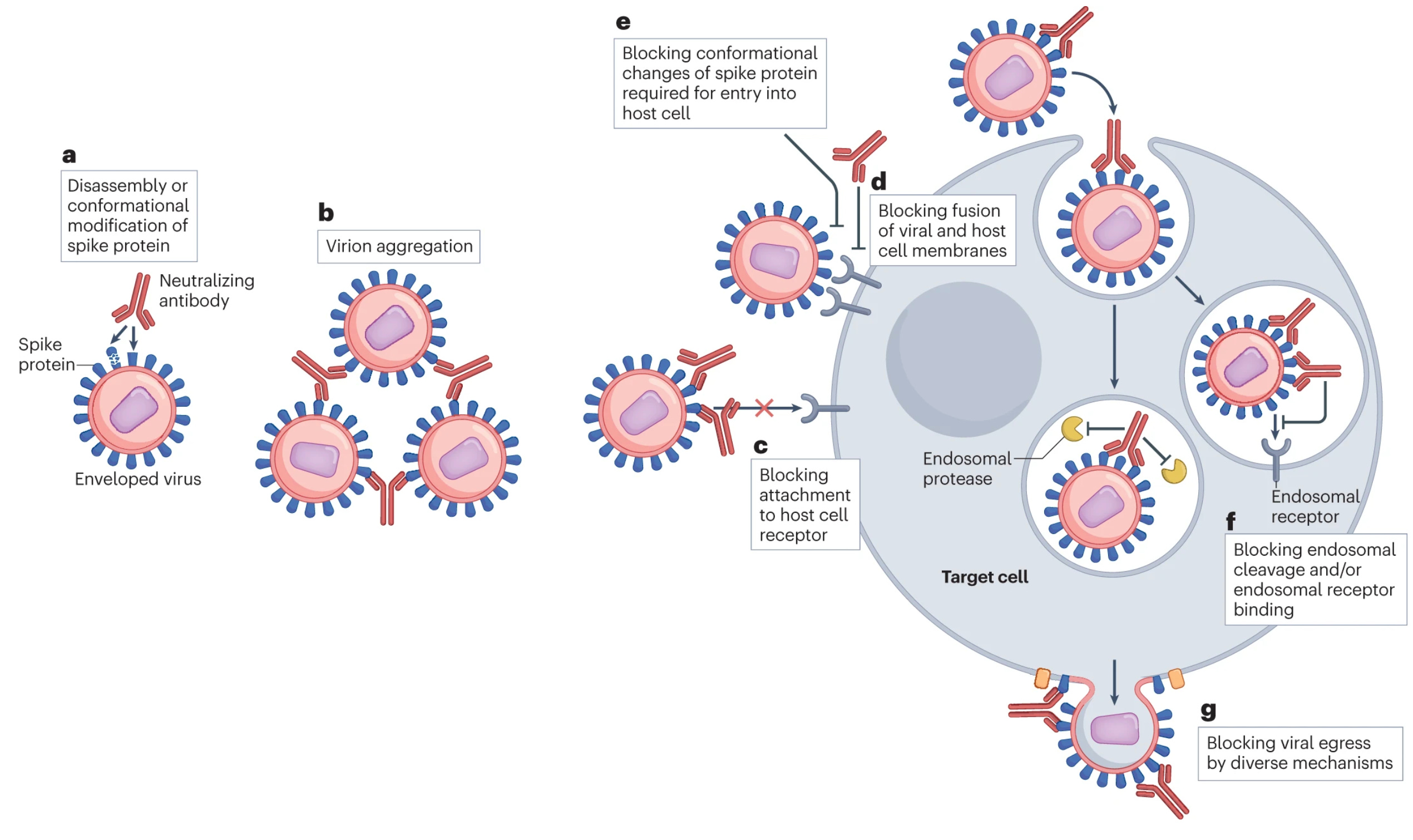
However, there is an entire world of antibody function beyond neutralization, and neutralization is not the be-all end-all of antibody function:

For this reason, even though a given antibody is neutralizing, it may still fail to provide protection if it is the wrong class or has the wrong modifications for a given context, and a non-neutralizing antibody can be protective if it is the right class or has the right modifications. The goal of vaccination and other interventions making use of antibodies is to ensure that these things match for their intended context. Given how many considerations go into the properties of antibodies and whether and when they are protective, it is hopefully becoming clearer why public health authorities were not willing to accept the presence of antibodies against SARS-CoV-2 alone as proof that individuals were protected against reinfection.
At times, people refer to the concept of sterilizing immunity. The term has several uses in the immunology literature, but in most contexts today, sterilizing immunity describes a state in which antibody titers are so high that an infection cannot occur because the microbe is neutralized before it can even begin replicating. In general this concept is mostly mythical: we do not have the ability to measure so precisely that we can conclude that not even a single viral particle (for example) has entered a cell (even PCR has a limit of detection). Another complicating factor is that the amount of antibody needed for sterilizing immunity will not readily reduce to a single value. Setting aside individual variation in each person’s immune system’s ability to respond to the antibodies, considerations of dose matter too. It takes far less antibody to stop a tiny amount of virus than it does to stop a much larger amount. If my exposure to an infectious person is brief, I can get away unscathed with a lower level of antibodies than if I have prolonged exposures (in fact, in some diseases, such as measles, cases tend to be more severe if the infection occurs from a household contact, which likely reflects the effect of a much bigger dose of virus). In the past, a declaration of sterilizing immunity was made when re-exposure to the microbe both failed to cause disease and failed to increase antibody titers, as the assumption was titers were high enough that the infection never took hold. In the era of qPCR where we can detect the presence of astronomically small numbers of microbes, this doesn’t hold up nearly as well.
There is a common misperception that vaccines only work if they provide sterilizing immunity, but in truth no vaccine or infection is shown to work like this, some are shown quite clearly not to work like this, and sterilizing immunity is not needed to control transmission of all microbes. This is made very clear in that a number of vaccines that are known not to provide sterilizing immunity have successfully eliminated multiple vaccine-preventable diseases:
In sum: antibodies are a critical component of the immune system (but not the entire immune system). They show incredible diversity and can steer the immune response in a given direction quite granularly depending on the context, and they lie at the heart of the protection mediated by vaccines.
Much like “all squares are rectangles but not all rectangles are squares,” all immunogens are antigens, but not all antigens are also immunogens. The classic example of an antigen that is not an immunogen is a small molecule known as a hapten. Because haptens are small, they can become attached to larger molecules known as carriers, and the complex of the hapten and the carrier can function as an immunogen. However, even though antibodies may recognize the hapten-carrier complex, the hapten alone will not provoke an immune response. Thus the hapten is an antigen but not an immunogen:
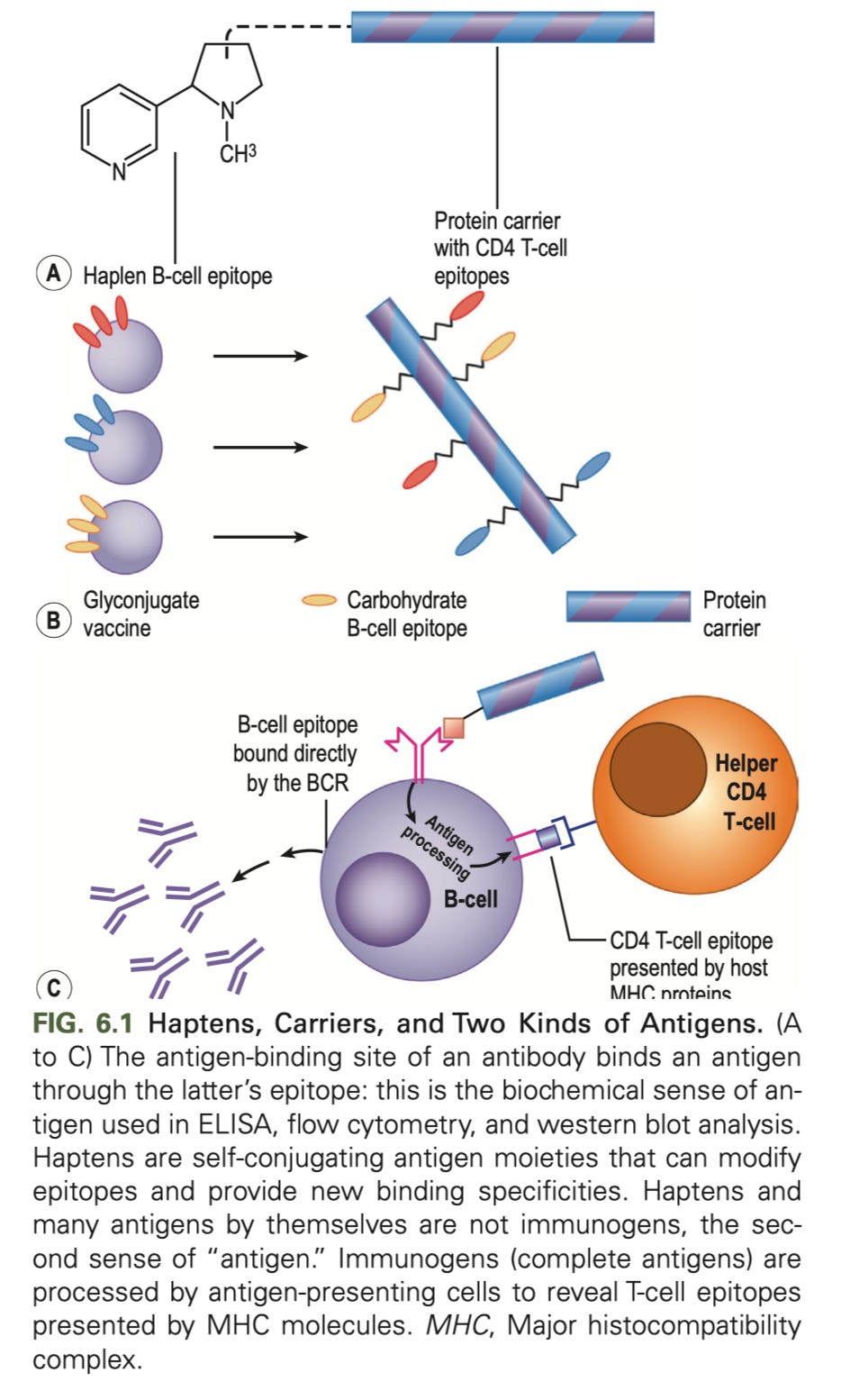
Is this distinction pedantic? Well, a bit. But as Pandley and colleagues write:
How scientists describe these terms can critically influence the types of hypotheses generated and experimental questions addressed. For example, knowledge of the viral antigenic sites recognized by Abs does not necessarily indicate which immunogenic structure initiates the production of Abs in the immunized host. Failure to differentiate between antigenicity and immunogenicity may lead to failures in developing synthetic peptide vaccines against viral diseases (37–39). How an immune response is measured has been a matter of great debate (40), and a lack of clarity regarding the difference between the two terms can further impede the understanding of advanced concepts associated with immunological cascades, such as haptens, adjuvants, allergens, tolerogens, etc.
Regardless, I wouldn’t hold it against people who don’t work in immunology if they default to “antigen” for everything.
T cells use T cell receptors (TCRs) to recognize antigen presented on MHC (major histocompatibility) proteins found on nearly all cells throughout the body. (Classic) MHC proteins however can only present peptides, and thus T cells can only respond to protein antigens- but there are some exceptions. For example, if the peptide fragment on the MHC contains an attached carbohydrate, the peptide might be presented with the carbohydrate together, and thus the TCR will recognize both. Additionally, there are unconventional subsets of T cells that recognize non-protein antigens. For example MAIT cells (mucosa-associated invariant T cells) recognize metabolites of vitamin B9 and B3 through the MR1 protein. iNKT (invariant Natural Killer T) cells recognize lipid antigens through presentation on CD1d.
You may have also heard of gammaglobulins (sometimes written as γ-globulins). These are so named because when you run electrophoresis (a technique that separates substances using their movement through an electric field) on serum proteins, you get a set of characteristic spikes in accordance with the charge of the proteins with (in order of most negative to most positive charge) albumin, α1, α2, β and γ spikes (incidentally, this is where the term prealbumin comes from- it’s the proteins in the serum electrophoresis spikes that come before albumin). However, you can resolve the components in even greater detail if you so desire:
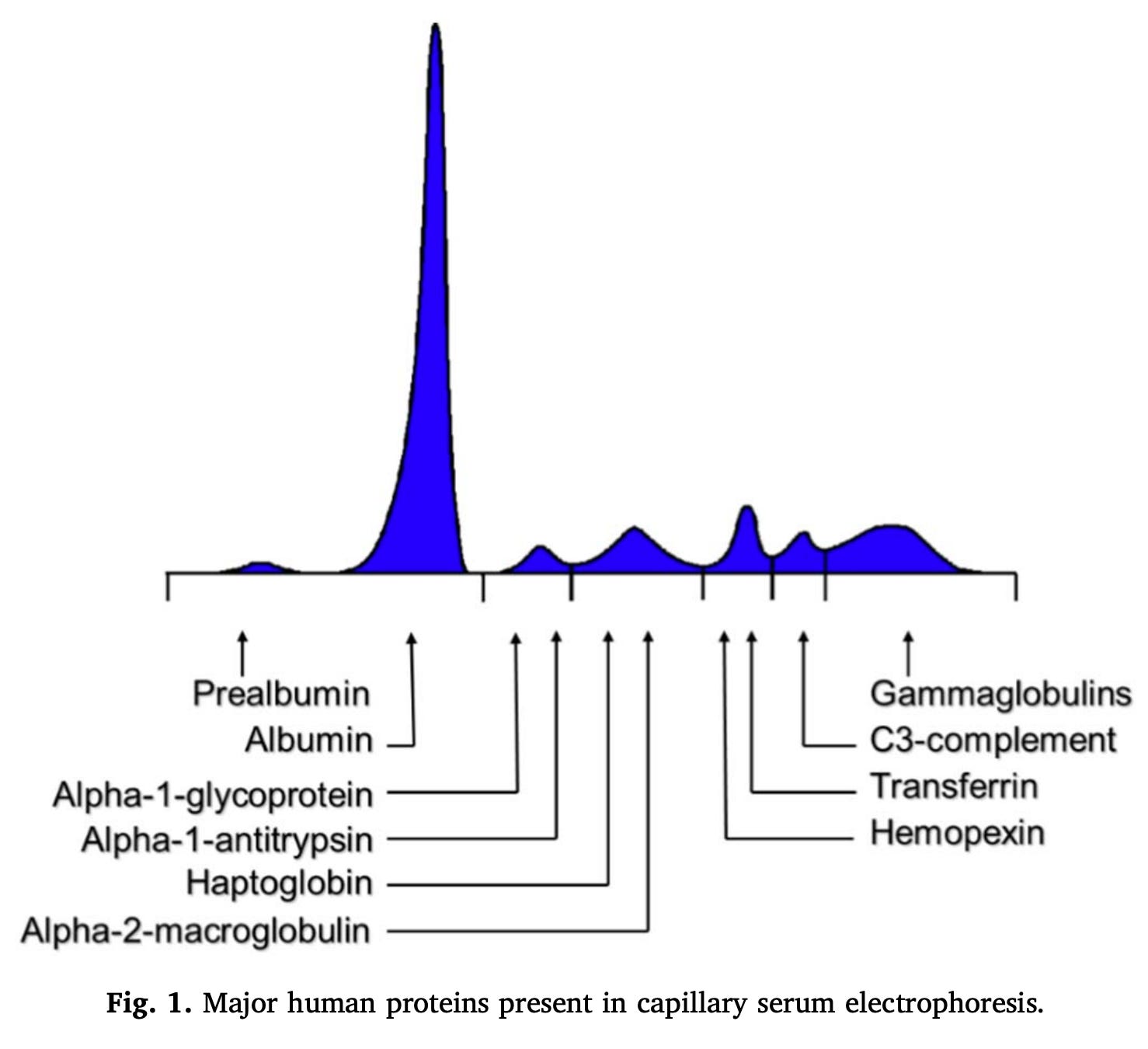
Gammaglobulins refer to the proteins found in this gamma region, which include immunoglobulins (antibodies). However, not all immunoglobulins are in the gamma peak and not all proteins in the gamma peak are immunoglobulins, so, formally, these terms are not synonyms. Despite this, there are conditions where there is inappropriate, excessive production of an antibody (or multiple antibodies) and these are still termed gammopathies, because there is a characteristic spike in the gamma fraction on serum protein electrophoresis seen with these conditions.
Biochemistry purists might be annoyed with me here because often models of protein binding derive from enzyme catalysis in which binding occurs not by a lock-and-key mechanism like I have alluded to here but through an induced-fit, in which the binding site evolves to minimize the free energy of the transition state, in general through maximizing binding interactions with it. However, it’s important to bear in mind that with antibodies we aren’t dealing with catalysis (unless we mean abzymes- taught as a classic in biochemistry courses to describe why enzyme active sites optimize binding to the transition state, and not the reactants), and evolution of the antibody is to bind the ligand, not the transition state. Indeed, germline antibodies may often exhibit induced-fit binding but as affinity maturation progresses adopt binding that more closely reflects the lock-and-key model. Nonetheless, the major reason I explain complementarity in terms of lock-and-key here is because this is I think easier for a layperson to conceptualize than induced-fit.
There are many different types of titers that can be measured and each one tells you different information. For example, in virology contexts, a neutralization titer is often measured by incubating dilutions of antibodies with the virus and observing what dilution allows the virus to regain the ability to infect cells. This can be further divided into microneutralization assays, plaque reduction neutralization assays, focus-forming assays, etc. depending on the details of the setup. Viruses can also be pseudotyped meaning their receptors are put onto a different virus that is more tractable to work with, such as if the virus is one that requires higher levels of biosafety than the lab performing the test is equipped for. These assays commonly report neutralization titers in terms of NT50s, the dilution of antibody needed to reduce the infectivity of the virus by 50%. A potential weakness of these neutralization assays however is that they often require knowledge of the virus’s receptor (at least if they are to be standardized) which is not always available.
Antibodies can also be evaluated simply for their ability to bind an antigen, without consideration of the downstream consequences. One common approach for this is an indirect ELISA, in which a plate is coated with the antigen and then the antibodies are applied in serial dilutions, after which a secondary antibody is added (in the case of an indirect ELISA) conjugated to an enzyme that will generate a signal when a reagent is added. The amount of signal will be proportional to the degree of binding by the antibody. This approach does not consider the orientation of antibodies and antigen, which can be important in some contexts (see the discussion on neutralization in the main text). However, ELISAs can be modified to give information about neutralization in some contexts. For example, there is a competition ELISA which allows for SARS-CoV-2’s spike protein receptor binding domain to (attempt to) bind ACE2 in the presence of antibodies:
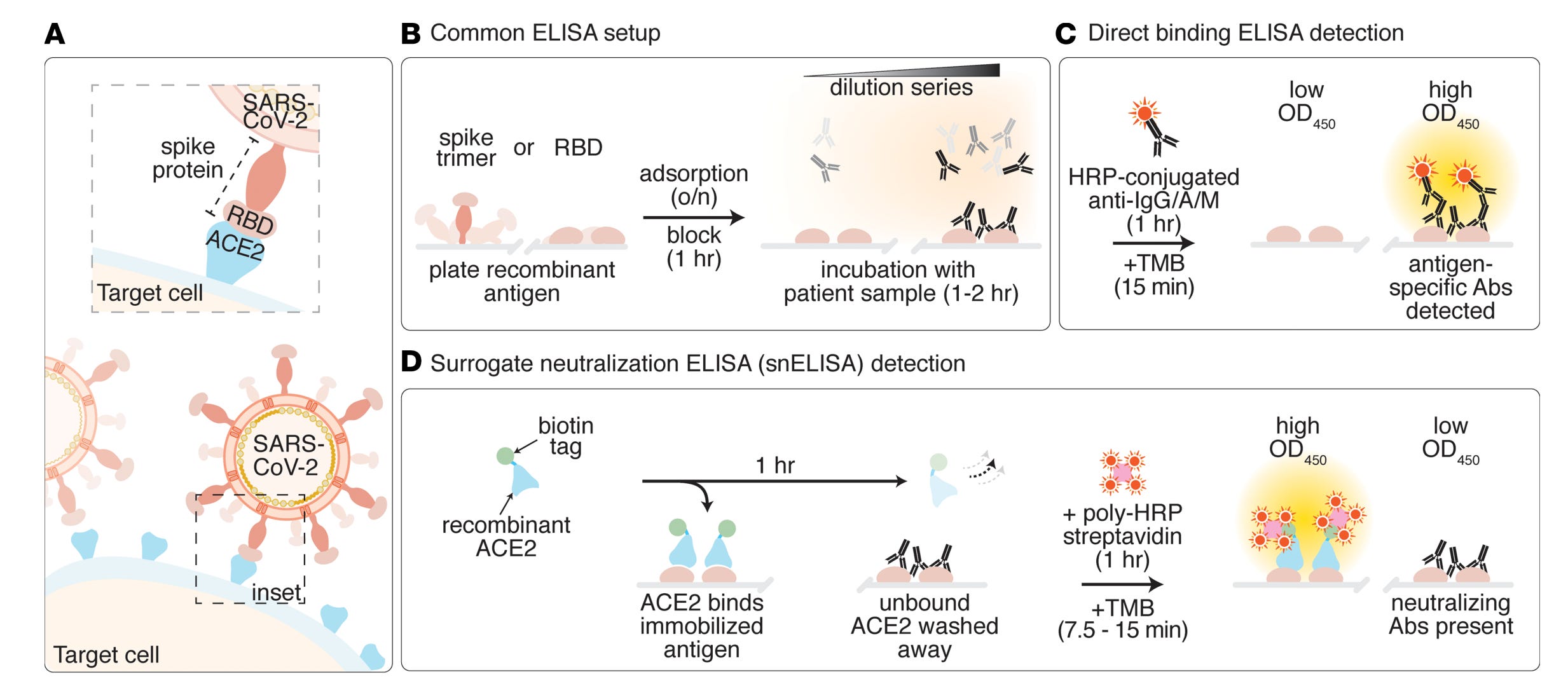
In the case of this specific approach, restricting to the RBD means that neutralizing antibodies outside RBD will not be detected.
In other contexts, the ability of antibodies to kill bacteria through complement fixation is critical for their protection (such as meningococcal disease), and so assays measuring serum bactericidal activity have also been developed. In these assays, the bacteria are incubated with serum antibodies and complement, and then cultured to determine at what dilution bacteria stop being culturable. The SBA titer is commonly reported as the dilution that gives 50% of the maximal bactericidal activity of the assay.
Some sources are really sloppy about their terminology and will call any short-lived plasma cell a plasmablast, but this is not correct. The most important difference between them is that plasmablasts have the capacity to divide (and in fact are rapidly proliferative) whereas plasma cells are terminally differentiated. It is also commonly taught that in contrast to plasma cells, plasmablasts have surface BCRs whereas plasma cells do not (consistent with the fact that the gene program that induces B cell differentiation into plasma cell changes splicing of the IgH gene so that the protein produced is destined for secretion rather than present on the surface; plasmablasts are at an earlier stage of differentiation and thus have not completely transitioned to having solely secreted BCRs). However, other work has argued that IgA and IgM plasma cells do have surface BCRs that are capable of signaling (but IgG plasma cells do not), and additional work has further shown that ligation of the BCRs on IgE plasma cells induces their death.
In this section, I did not distinguish functionally between naive and memory B cells, but this distinction is important. Specifically, naive B cells require much more costimulation in addition to antigen to respond than memory B cells. This allows memory B cells to respond much more quickly than naive B cells, and the response is consistently at a much higher magnitude.
Memory B cells (MBCs) and (long-lived) plasma cells (LLPCs) are often said to comprise 2 walls of immunity:

Plasma cells secrete antibody constitutively independent of stimulus (although TLR agonism is shown to increase their rate of secretion). This ensures a non-zero level of antibody persists throughout the lifetime of the plasma cell (which can indeed be the entire lifetime of the organism). However, pathogens evolve and mutate, allowing them to escape our antibodies. Making things worse: plasma cells lack the ability to divide or undergo affinity maturation to adapt to these variants. However, memory B cells are capable of rapid differentiation into antibody-secreting cells, a portion of which will become long-lived plasma cells. If a novel variant emerges, the memory B cell compartment is highly adapted to rapidly generate antibodies that cover that variant.
Presently, we cannot rationally control to what extent a given immune response produces plasma cells vs MBCs, and we furthermore have not identified means of ensuring that the plasma cells that result are long-lived.
Below is a summary of the IgG Fc receptors (i.e. Fcγ receptors) seen in humans from Paul’s Fundamental Immunology 8th Edition modified from Figure 33.1A:
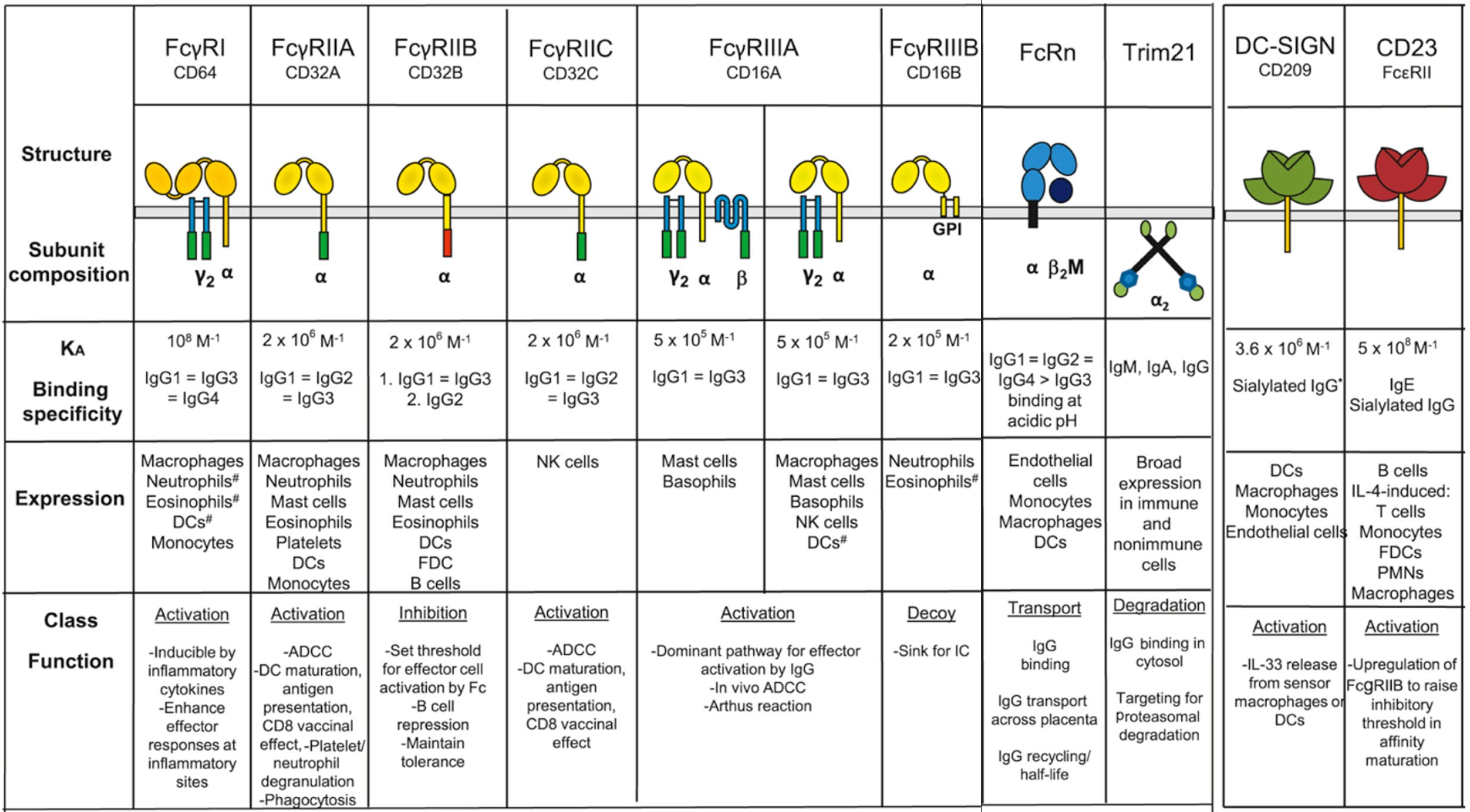
Much (in fact, most) of the functions of IgG are exerted not by the antibody itself but by its interactions with FcγR-bearing cells. These receptors are divided into type I and type II based on their structural features (type I receptors are immunoglobulin receptor superfamily members that bind antibody in a 1:1 antibody:receptor stoichiometry, while type II receptors are C-type lectins that exclusively bind the closed conformation of IgG in a 2:1 receptor:antibody stoichiometry) and also classified as activating or inhibitory receptors depending on the outcome of their engagement. In general, the type I receptors are all activating except FcγRIIb, which is unique in that it has an ITIM in its cytoplasmic tail whereas the others all have ITAMs. Type II receptors by contrast tend to ultimately have inhibitory functions, usually through inducing or enhancing the action of FcγRIIb.
Activating receptors can induce FcγR-bearing cells to kill cells to which antibodies are bound (through many different mechanisms), produce cytokines or chemokines (to recruit other components of the immune system), to enhance expression of costimulatory molecules (to help activate B and T cells to respond to the threat), to process and present antigen, and more. In turn, the inhibitory receptors suppress these behaviors. Type II receptors help to induce expression of the only inhibitory type I FcγR- FcγRIIb to suppress these responses.
In general, it is thought that meaningful engagement of most of the Fc receptors (except for FcγRI) cannot occur with antibodies alone but rather requires the antibodies to be tethered together by the antigen they in immune complexes- thus inflammatory responses elicited by antibodies for the most part require that the (multivalent) antigen be present within the body to occur. This is because the binding of the FcγRs is not tight enough to hold onto individual antibodies (note association constant values). The different FcγRs have different affinities depending on the type of antibody (IgG1 vs. IgG2 vs. IgG3 vs. IgG4) and structural features of the antibodies.
Another key structural feature of antibodies that determines their function is a glycan located at N297 within CH2 of IgG:

These glycans can cause the IgG molecule to shift into an open or closed conformation, enabling or hindering binding to Fc receptors:

For example, absence of fucose (afucosylation) greatly enhances binding to FcγRIIIa, which allows these antibodies to be extremely potent inducers of inflammation. In contrast, sialylation of this glycan imparts the antibody with anti-inflammatory properties. Both of these glycoforms are relatively rare components of overall IgG, but can have important functional consequences. Intravenous immunoglobulin (IVIg) can be given as replacement therapy for those who have B cell deficiencies, but high dose IVIg is often used in the treatment of autoimmune diseases for its anti-inflammatory effects. The anti-inflammatory effect of IVIg is thought to come specifically from the sialylated fraction of IgG (although some components are likely independent- for example, FcRN controls the half-life of IgG and can be saturated to clear pathogenic antibodies through high dose IVIg, regardless of the glycoform). The mechanisms underlying that are more complex as there is dispute on the biochemical details regarding which receptors are bound by sialylated IgG. In general, the presence of sialic acid at the glycan is thought to enhance binding to type 2 Fc receptors, which subsequently enhance expression of FcγRIIB to increase the activation threshold for cells. However, some have also reported that sialylated IgG has enhanced affinity for FcγRIIB. In both cases, there are papers reporting no binding to sialylated IgG by type II receptors or no enhanced binding to FcγRIIB. However, the anti-inflammatory effects of sialylated IgG (and high dose IVIg) in vivo is a robust finding that is highly reproducible and shown to depend in vivo on FcγRIIB and the type II Fc receptor SIGN-R1 (the mouse equivalent of DC-SIGN), DCIR, and CD23.
In addition to IgG receptors, there are receptors for other antibody classes (also modified from Figure 33.1A):
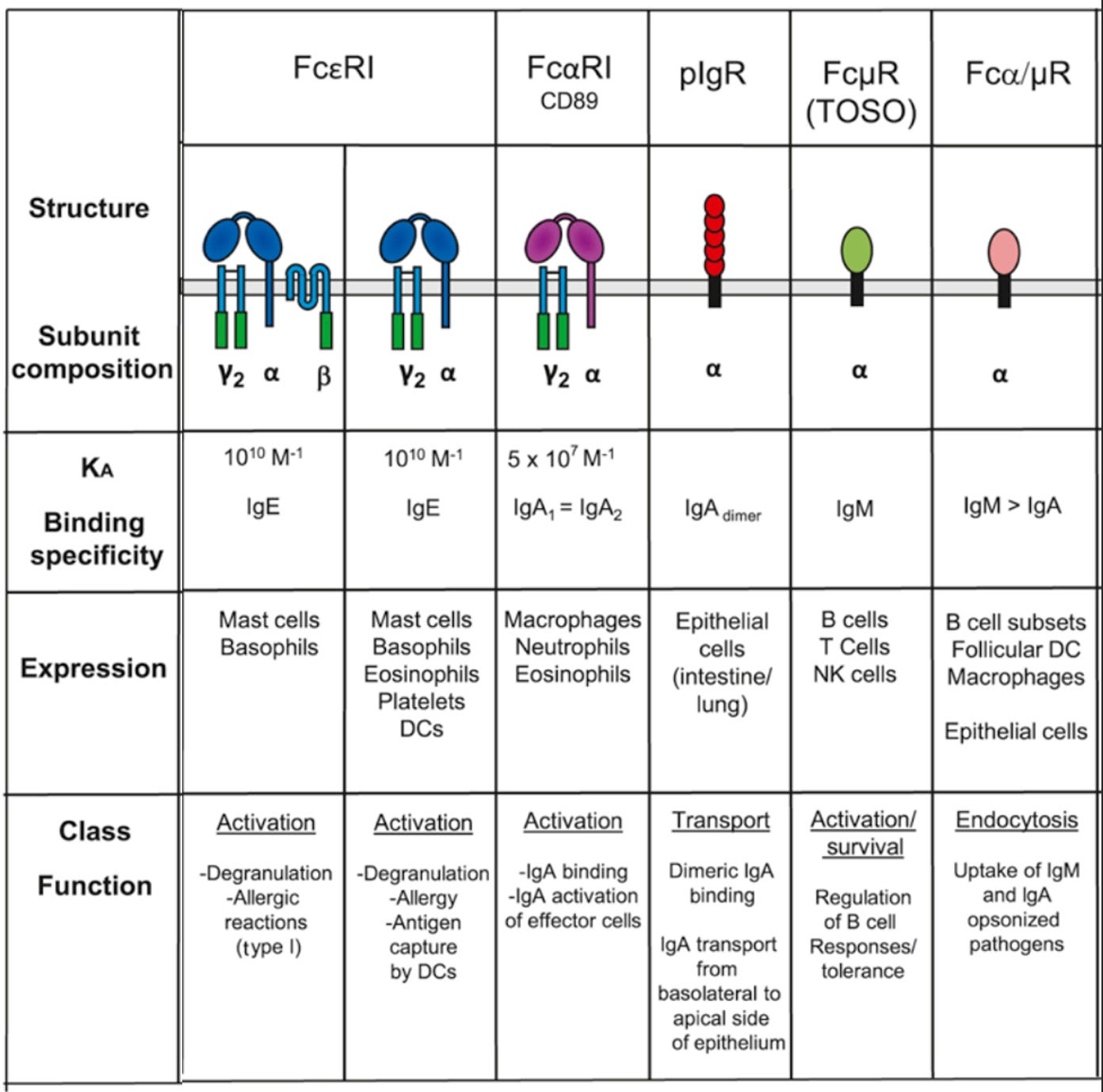
The specific contributions of these receptors to human health is not as well understood as for the FcγRs.
It is worth providing some additional detail on how the different classes and subclasses of antibodies work.
All B cells initially make IgM, followed by a stage in which they express both IgM and IgD, and then generally undergo class switching to produce IgG, IgE, or IgA antibodies. IgM antibodies exist mainly as complexes of 5 IgM proteins linked together by a protein known as a J chain, but have a very short half-life. In general, IgM antibodies tend to have low affinity as they are produced by newly minted antibody-secreting cells (and those ASCs have to avoid cytokines that drive class switching), but compensate for this through having 10 paratopes with which to bind antibody. IgD antibodies are a very minor fraction of overall antibodies made and seem to mainly be important for activating cells known as basophils but their function otherwise (and its contributions to human health) is not entirely clear (deficiency of IgD is very rare and doesn’t have any clear health consequences). IgE antibodies are mainly important for activating mast cells, making them the drivers of allergies, while also playing important roles in the defense against parasites. IgA antibodies are the major antibodies at most mucosal surfaces, also typically found in the form of secretory IgA, which includes two IgA molecules linked by a J chain like IgM as well as a secretory component made from the pIgR (polymeric immunoglobulin receptor) protein, serving to protect them from being destroyed by enzymes. Humans further have two subclasses of IgA: IgA1 and IgA2. IgA2 is more resistant to degradation by enzymes that degrade IgA1 and is reported to be more effective in inducing inflammation; however, the study of the functions of IgA has been hampered by the fact that mice lack an Fcα receptor (whereas humans do have one). Interestingly, despite the fact that IgA is the most abundant antibody at mucosal surfaces by far, selective IgA deficiency is the most common primary immunodeficiency and causes no symptoms in the majority of people.
IgG antibodies are the best-studied and merit additional detail:
IgG1: Accounts for 60-75% of all IgG antibodies in the serum and can have either anti- or pro-inflammatory functions depending on the context (especially glycosylation). This is the best-studied of the subclasses because it represents the bulk of all IgG activity. Deficiency of IgG1 generally reproduces the effect of a combined IgG deficiency because it represents the bulk of all IgG activity in the body.
IgG2: Mainly induced in response to polysaccharide antigens, such as the capsules of bacteria. IgG2 can exist in 3 different forms depending on how disulfide bonds on it are rearranged called A, B, or A/B; in general, we do not need to concern ourselves with this but in the context of therapies, designing IgG2 in a particular conformation can be valuable. IgG2 is a poor activator of complement and exhibits poor binding to the type I FcγRs and no substantial binding to FcγRI. It is, however, more resistant to proteases that other subclasses are sensitive to. Normally accounts for 20-30% of all serum IgG.
IgG3: The most potently inflammatory of the antibody subclasses and the most variable across the population due to a number of variants (Gm allotypes). It tends to be made early on in immune responses, and is thought to buy time for the rest of the immune response to get going as it activates the other components of the immune system. Because it has a very long and flexible hinge, it appears to be better at binding 2 antigens at the same time (called bivalent binding) than other IgG subclasses, making it potentially attractive for antiviral antibody therapeutics. It has the shortest half-life of all the IgG antibodies at about 7 days in adults (the rest have a half-life of about 21 days; some people have a genetic variant called an allotype in which arginine at position 435 is mutated to histidine, R435H, that gives it the same half-life as other IgG antibodies but there are suggestions that this is associated with a higher risk of autoimmune disease). Normally makes up about 3-8% of antibodies in the serum.
IgG4: An antibody associated with repeated and/or chronic exposures to an antigen, especially in the setting of allergen immunotherapy or in antibodies against antibody medications. While not actively anti-inflammatory, it can reduce inflammation by competing with other antibodies for binding sites on an antigen and thus not allowing them to interact with low-affinity Fc receptors (which is especially important in allergen immunotherapy as this helps to prevent anaphylaxis). IgG4 is unique in that it can split into half-mers through a process called Fab arm exchange where instead of having 2 binding sites for antigen, it has 1. These IgG4 can also recombine with other IgG4 to form IgG4 molecules that are specific to 2 different antigens. Because IgG4 undergoes this process, it has reduced ability to form immune complexes (these require multiple binding sites on both the antibody and antigen) and therefore reduced ability to signal through Fc receptors. Normally makes up about 1-4% of all IgG in the serum.
One key aspect of antibody function in vivo is the A/I ratio- a metric for how well a given IgG subclass binds the activating to the inhibitory receptors:

Lower values indicate that an antibody is especially sensitive to the effect of FcγRIIB. This, however, depends on the variant of the Fc receptors as well:
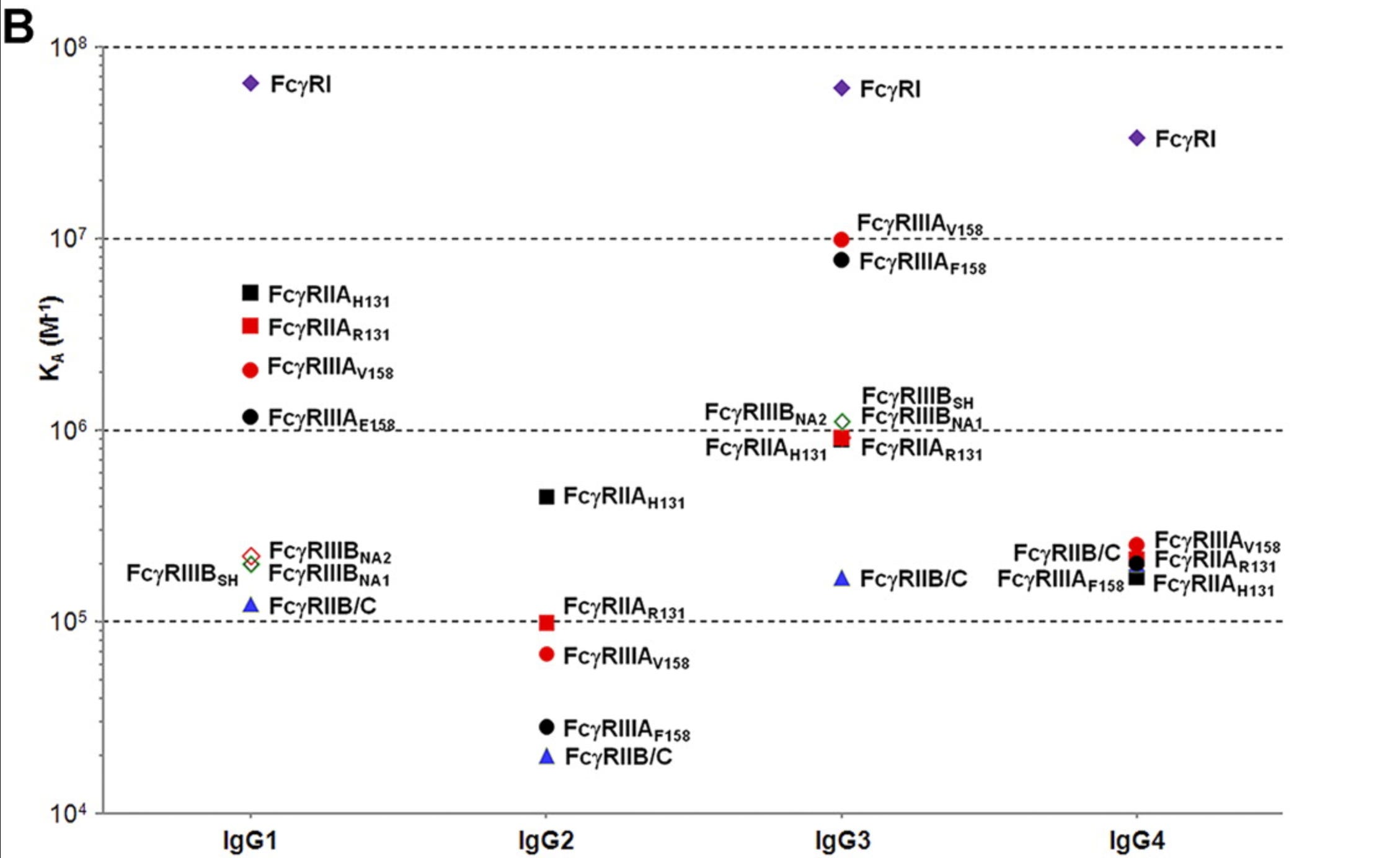
FcγRIIIA (CD16A) has a high-affinity variant (V158) and low-affinity variant (F158), and depending on this, the A/I ratio for human IgG1 can be either 1 or 13. If one expresses the low affinity FcγRIIIA, this actually gives IgG2 a higher A/I ratio than IgG1 at 1.5, but only 3.5 with the high-affinity variant, which is less than the 13 seen with IgG1. IgG3 invariably has the highest A/I ratio by a large margin. Caution is needed however in considering these metrics because in addition to subclass, A/I ratio is sensitive to IgG glycosylation. For example, afucosylated IgG has been shown to bind with 50- to 100-fold greater affinity to FcγRIIIA compared with fucosylated, which means a 50- to 100-fold increase in the A/I ratio, a finding that has critical importance in both immunotherapy and in understanding the immunopathogenesis of some infectious (dengue and COVID in particular).
Another factor in the binding and function of antibodies is the glycosylation of the Fc receptor in question. Specifically, FcγRIIIA shows differences in affinity for human IgG1 depending on its glycans, and deglycosylated CD16 has enhanced binding to IgG1.
While antibodies are usually regarded as inhibiting what they bind to, agonistic antibodies which activate the target of their binding also exist. For example, Graves’ disease is a condition in which antibodies target the TSH (thyroid stimulating hormone) receptor and activate it, resulting in enhanced, dysregulated production of thyroid hormone and causing hyperthyroidism, as well as orbitopathy, where there is proliferation of fibroblasts and adipocytes in the eye, causing swelling. This is visually summarized and discussed in detail here:

In some contexts however, this agonism is a goal, rather than a pathology. For example, antibodies that can activate costimulatory molecules that cause immune activation (like CD40) can be very valuable in cancer immunotherapy where the goal is to generate an immune response against the tumor. For clinical effectiveness, these antibodies typically require engagement with the inhibitory Fc receptor FcγRIIB, presumably because engaging other (activating) Fc receptors would mean loss of the agonist antibody target cell by inducing its killing. This also allows for clustering of the agonist target on the surface to achieve potent signaling. However, work on IgG2 suggests that this dependence is not absolute. Specifically, IgG2B isomer is able to exert agonist actions without needing to engage with FcγRIIB. Because A, B, and A/B shift with the redox state of the environment, when applied therapeutically, IgG2 can be modified to have a locked B form.
Affinity maturation occurs through a process known as somatic hypermutation. B cells express an enzyme known as activation-induced cytidine deaminase (AID- AICD means something else in immunology and was discovered first), which performs a deamination reaction that converts deoxycytidine in the DNA (yes DNA) into deoxyuridine in regions of the genome that are being actively transcribed. Because uridine is not supposed to be present in the DNA ever, this induces DNA repair mechanisms which will randomly modify the DNA encoding the immunoglobulin (and potentially elsewhere, but this is rarer than the immunoglobulin genes). AID targets DNA that is actively being transcribed and thus has a strong preference for the immunoglobulin locus in B cells, but it can target potentially any actively transcribed region, which can (rarely) cause lymphomas.

Naturally, the vast majority of these mutations are useless or harmful. To address this, we have the cyclic re-entry model. This occurs within germinal centers. The germinal center is divided into a dark zone and a light zone.
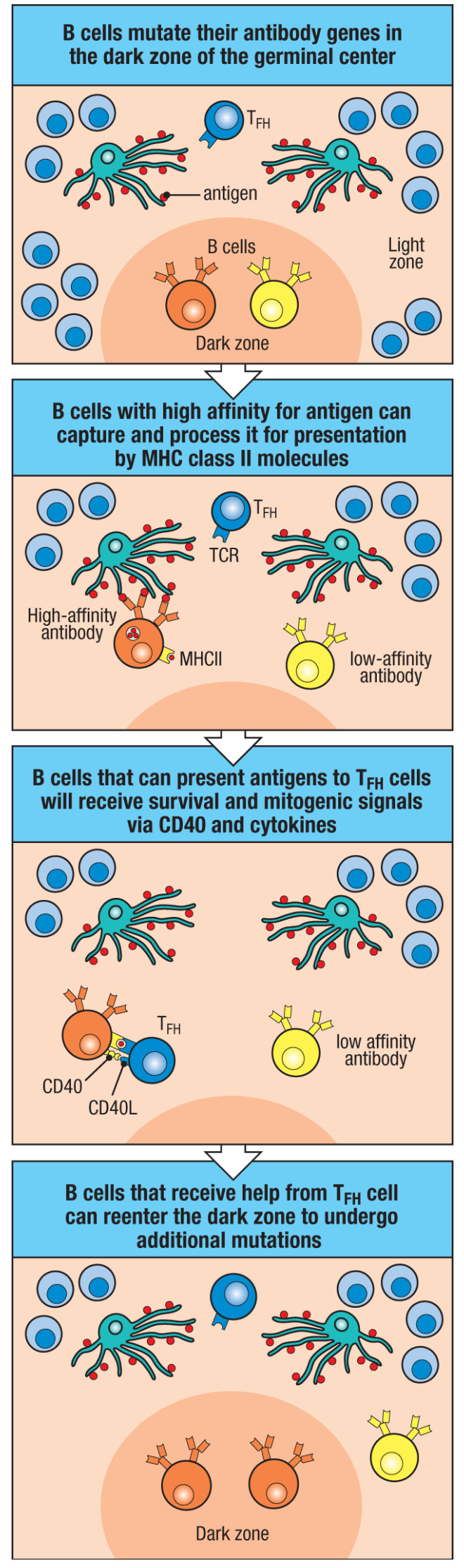
In essence, B cells in the dark zone will take their new mutated BCR and attempt to use it to bind and endocytose antigen that has been gathered by a follicular dendritic cell through CXCR4-dependent migration. Most of the time, the mutated antibody is unhelpful and the B cell will not be able to take up the antigen (or at least, it will be outcompeted by B cells who can take it up more effectively), so it dies from a lack of survival signals. If it succeeds in making an antibody that binds the antigen, the B cell will present the antigen to a T follicular helper (TFH) cell, and the TFH cell will supply the B cell with costimulatory signals that will promote its survival and differentiation. The B cell can repeat this process for many rounds, iteratively improving the quality of the antibody and producing antibody-secreting cells along the way.
For class switching, AID is also required, but it is now believed that it occurs mainly outside the germinal centers. Class switching occurs when there are double-stranded breaks in the process of repairing AID-induced lesions, resulting in excision of the intervening DNA.

Upstream of each constant region within the introns lie switch (S) regions. These S introns are spliced out of the nascent RNA transcript but form G-quadruplex structures that can bind AID and recruit it to that particular switch region to induce deamination, and eventually cause a double-stranded break that enables the class switching as above.
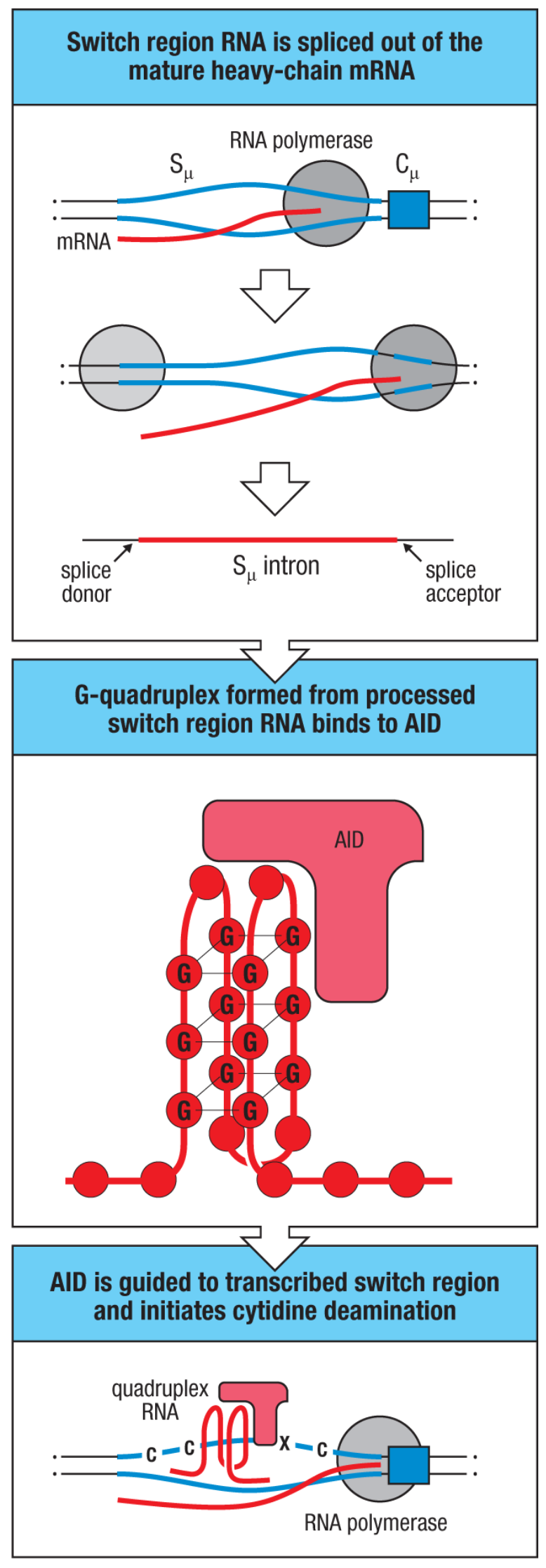
Class switching is regulated by cytokines from TFH cells elicited in the course of the immune responses. Different cytokines are produced in response to different types of immunological threats, so this allows for selection of antibodies that have the relevant effector function profile to combat the threat in question. The cytokines work by inducing transcription of a particular switch region through inducing transcription of the I regions that lie just upstream of the S regions. Additionally, class switch recombination can occur in a sequential manner provided that the relevant constant regions exist in the DNA of the B cell in question (i.e., if you have already spliced out the constant region, you cannot, in general, class switch to that region). This is believed to be important in allergy wherein there is initially a class switch to IgG1 which gains affinity for the allergen, and then a second switch occurs to IgE, enabling the allergic response (although the role of such sequential class switching in the development of allergy has been questioned in recent work). Similarly, it has been found that IgG4 can readily class switch into IgA2 in humans.
Cytokines that induce class switching may also have other sources. For example, because T cell receptors can only recognize peptides presented on MHC, they cannot provide help with polysaccharide antigens (some unique circumstances can arise however). However, we can still observe class switching against polysaccharide antigens even without conjugation to a protein:

This is thought to be from cytokines produced by dendritic cells, especially BAFF.
While you do have (very rare) B cells that express only IgD, IgD is generally expressed not through class switching but by alternative splicing in the B cell where the mRNA of the heavy chain uses the D region exon instead of the M region. Because IgD is so rare, it is generally only thought of as a marker of B cell maturity as all B cells must express a surface IgD in their development. However, more recent work has found that IgD plasma cells are enriched at the mucosae and play a role in defenses therein.
While the hyperlinked example is a therapeutic one, these antibodies can occur physiologically (but there isn’t a guarantee that they won’t affect receptor function if they do occur). A model for this phenomenon according to immune network theory is shown below:

Initially, an antibody is generated against a virus envelope protein (for example). This antibody may (by happenstance) recapitulate some of the structural features of the receptor for the virus that binds the envelope protein. Then, an antibody is induced against the first antibody’s Fab region (this is known as an anti-idiotypic antibody). This antibody may exhibit crossreactivity with the viral receptor in addition to the original anti-envelope antibody. In the context of an infection, this could help to curb infection by the virus, but could also result in pathology depending on the nature of the receptor. It has been proposed that this is a mechanism by which antibodies against ACE2 arise and that these antibodies may contribute to long COVID or more severe manifestations of COVID acutely. In the same vein, antibodies against the spike protein with the ability to catalyze the conversion of angiotensin II (or angiotensinogen) to ang(1-7) have also been reported, although what effect these may have on health (if any) is not currently known. Nonetheless, this is not proven, particularly with long COVID, which likely represents a syndrome with many endotypes rather than a singular discrete disease entity.
Thanks Dr N . Such a rich and complex subject . What a class of macromolecule, right ??? Amazing . I really appreciate the way you have boiled it down for us practicing docs . I know it is hard to do that - especially when textbooks on the subject are , well , easily hundreds of pages . Plus a nice job mentioning T cells and how they relate to antibodies . You mentioned them but did not go off on a T. Cell tangent . Finally , and maybe the best part is the discussion of neutralization . Not being in research I have puzzled over that concept many times. You explained it as well as I have ever heard. Kudos.