



[Imported Post from December 5, 2020] What is Antibody-Dependent Enhancement (ADE)?

Editorial note: while this post is old, it is still basically correct and I think suffices as a reference for someone seeking to understand ADE. Regarding SARS-CoV-2, to date (June 14, 2024), no convincing evidence has emerged to support that ADE is an important contributor to the disease caused by this virus. I am uploading this here as part of my efforts to consolidate everything onto a single place.
The short version: Antibody-dependent enhancement (ADE) is a rare phenomenon basically limited to dengue and a few other infections where the presence of antibodies makes a disease worse. The antibodies can come from anywhere: prior infection, passive immunization (e.g. convalescent plasma), or vaccines. This was a concern for the development of COVID-19 vaccines. The current evidence we have at this point in almost a year of a pandemic is that this concern is not reasonable. Convalescent plasma has been used extensively throughout this pandemic and if ADE were significant, we would have evidence by now.
Part I: The Fundamentals
A few times on my blog, I’ve discussed antibody-dependent enhancement (ADE) but I’m seeing it come up more and more as we approach EUA and eventually approval for COVID-19 vaccines, so I thought it would be helpful for people to have a separate post discussing it in some depth.
ADE refers to a phenomenon wherein the existence of antibodies to a given infectious agent results in more severe disease. On its face, it should sound unusual. You might remember from your high school science classes that after an infection you generate memory cells, and these allow for a more rapid and robust response if you encounter the pathogen again that should keep you from getting sick. You would be right- it is unusual. It is so unusual that there are very few examples of it occurring, and very specific conditions have to be met for it to occur. In general there is a good deal of evidence that ADE occurs with Dengue infections, some evidence it can occur with Zika infection, some evidence it can occur in Ebola, and that’s honestly about it (it has been documented for other infections but there hasn’t been evidence that it occurs in a way that is significant for the people’s health). ADE was a significant concern in the design of a SARS-CoV-2 vaccine, and indeed for the possibility of reinfection with SARS-CoV-2, because there was some evidence that it could occur for other coronavirus infections (but this was based on preclinical models- animals and cell cultures- which are NOT predictive of ADE in humans- elaborated upon in the next section- but do signal that it’s something that needs attention throughout the clinical trial process), and we didn’t know in advance that it wouldn’t occur for COVID-19 or vaccines against SARS-CoV-2.
So, naturally the big question is, is there ADE happening with COVID-19 infections or vaccines, and the answer is: almost certainly not. The reason we can say that is because of the widespread use of convalescent plasma in COVID-19 patients (emphasis mine):
The administration of passive antibodies could also reveal whether antibodies predispose to ADE of disease…
The evidence that COVID-19 does not worsen after treatment with plasma from convalescent patients has been substantially reinforced by a study of 20,000 patients who were severely ill with the disease, showing an adverse event incidence of 1–3% [81]. If further substantiated, these findings will markedly diminish the concern that clinically relevant amplification of infection, release of immunopathogenic cytokines or immune-complex deposition in the presence of a high viral load is mediated by SARS-CoV-2 antibody-dependent mechanisms [82,83].
I should add that Arvin et al.’s Perspective from which the above quote is taken is the best literature I have read on the question of whether or not ADE is a reasonable concern with respect to all things COVID-19, so I strongly recommend that for anyone seeking more details.
This suggests that the chances of ADE occurring for anyone who has had COVID-19 or receives a COVID-19 vaccine are either very rare or nonexistent. So, the simple answer of whether or not you should worry about ADE for your COVID-19 vaccines is: no. Surveillance should be continued for any evidence of ADE, but odds are if it were at all a significant problem, we would have seen it by now, especially since we have documented reinfections of SARS-CoV-2.
If this satisfies your concerns, you can stop here. If you want to know exactly how this happens… well you will be disappointed (because no one is certain), but I discuss some ideas in the next part.
Part II: The Whole Story

So you might be wondering at this point how and why this sort of thing happens, because it is weird. No one is entirely sure (at least regarding the details). The best-characterized example occurs with dengue, so I will focus on that one. Dengue is a mosquito-borne virus that comes in 4 serotypes (strains). A second infection with dengue can sometimes produce a condition called dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS), particularly if it is with a different strain of virus than the first dengue infection. Current dogma for how this occurs goes something like this: after an initial infection, antibodies specific to the antigens of that dengue strain develop develop, and these antibodies can bind and neutralize viral particles to prevent their entry into cells. Then, a group of receptors called Fc (for “fragment crystallizable,” the part of the antibody that’s like the trunk of the “Y”) receptors can recognize antibodies and lyse bound virus. It is thought that when infected with a different strain of dengue, antibodies from a prior infection get recalled. These antibodies are able to bind, but cannot effectively neutralize dengue virions (sometimes called subneutralizing antibodies). The dengue virions get taken up by cells bearing Fc receptors, and then can initiate a productive infection, spreading to cells they previously would not have, especially macrophages. The macrophages seem to become viral replication factories, which enhances inflammatory responses triggered by the dengue viruses. This leads to increased vascular permeability that produces shock (a state where the entire body’s oxygen demands are not being met, generally because of inadequate blood flow, like if many vessels suddenly dilate and there’s a significant drop in blood pressure, called distributive shock). Another factor that implicates antibodies in the condition is its occurrence in infants. IgG antibodies can be passed across the placenta from mother to fetus, and persist for the first few months of life. If low avidity (subneutralizing) antibodies are present in an infant that then contracts dengue, it can produce DHF/DSS. This has never been observed in children born to mothers who lack antibodies to dengue. Indeed, viral loads of organisms who have subneutralizing antibodies are higher than those who are immunologically naive and have no antibodies. This example also really highlights the immunological concept of avidity. In biochemistry, we care about how well various molecules can bind to one another, which is known as affinity. In immunology, we are much more concerned with avidity- the sum of all binding interactions. This is important because administration of antibodies known to be neutralizing at concentrations that are subneutralizing reproduces the effect of DHF/DSS, which indicates that avidity, and not affinity, is the key problem here. Why would that be? Well, consider a virus for a moment. You have all sorts of structural proteins on your surface including a fusion protein that lets you get into cells. All you need is exposure of the fusion protein to its receptor to get in. If there aren’t enough antibodies to block that from happening, you can still enter cells despite whatever neutralizing antibodies have bound. But, all antibodies can be detected through the network of Fc receptors. That means that low titers of a neutralizing antibody can still lead to enhanced tropism (the virus can infect cells it ordinarily doesn’t). Hence avidity matters much more than affinity.
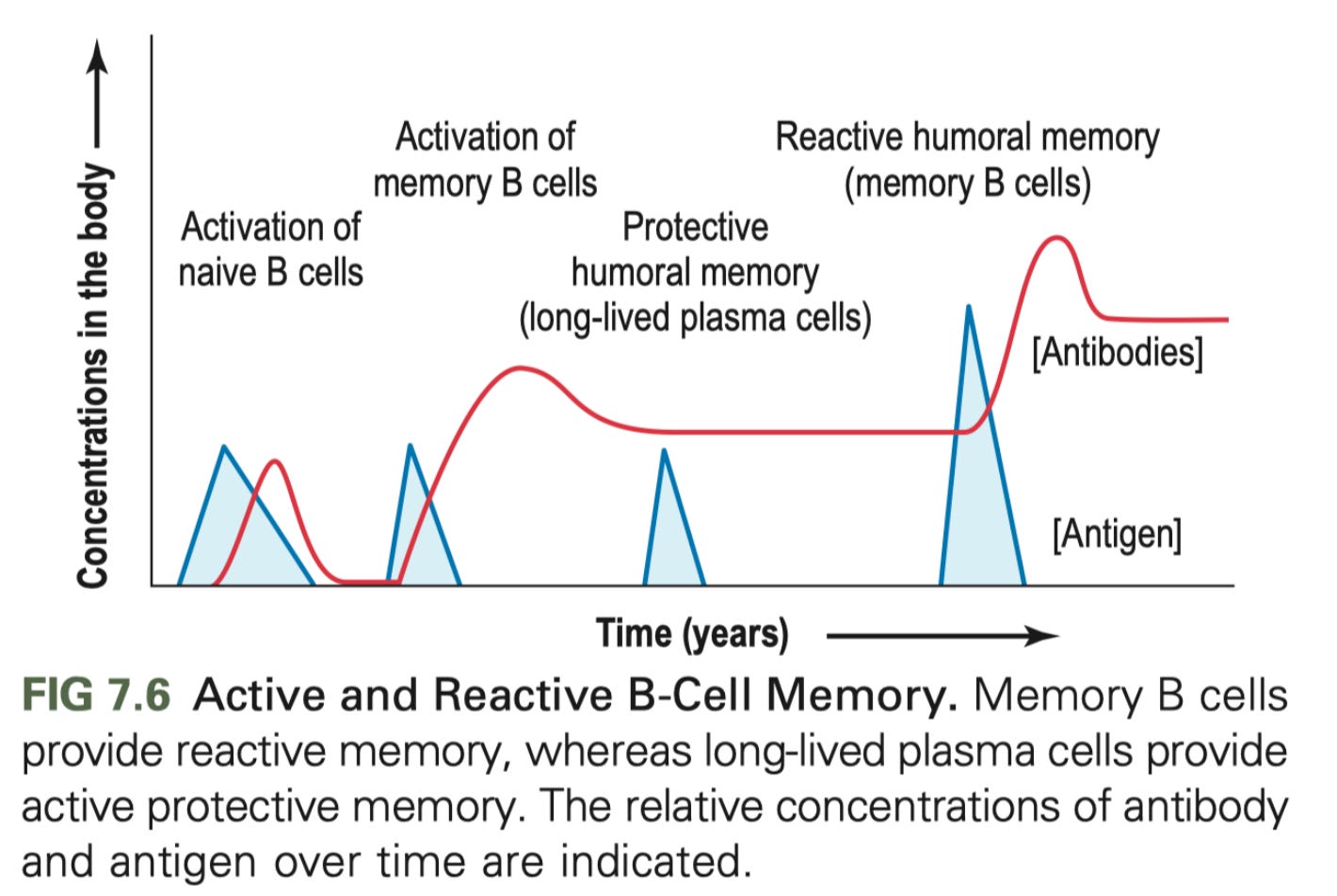
The fact that ADE can occur when titers drop may seem somewhat disconcerting, especially in light of the hysteria of waning antibodies a short time after infection. However, the decline of titers is normal following an adaptive immune response because some of the B cell clones (called plasmablasts) die off as they are unneeded, as some become long-lived memory B cells that can be recalled upon re-encounter with the antigen they are specific to rapidly divide into many B cell clones that all secrete antibody. You also make long-lived plasma cells which constantly secrete huge quantities of antibody. The diagram on the right also shows that after a challenge of antigen in the body, there is a rise and then a rapid decline in the levels of antibodies. This is known as the anamnestic response (an = without, amnestic = amnesia; i.e. memory). It’s normal and appropriate that many of these plasmablast clones die off- this doesn’t occur in some lymphomas, which needless to say is… very bad. Also as Professor Barker has eloquently explained before on Immune and TWiV, we would all be giant lymph nodes if this didn’t happen as we just accumulated more and more B cells (indeed, the swelling of your glands, which are lymph nodes- called lymphadenopathy- is thought to occur from an influx of large numbers of B and T cells which undergo rapid replication). It is probable that for a serotypically homogenous virus (by which I mean, one strain), there is very limited potential for ADE to start with, and SARS-CoV-2 fits this descriptor. Furthermore, as far as vaccines, Moderna has reported preliminarily that after a slight decline in titers initially, the levels of antibodies have remained steady from their vaccine in Phase 1 participants for months, consistent with what would be expected from a robust, anamnestic response.
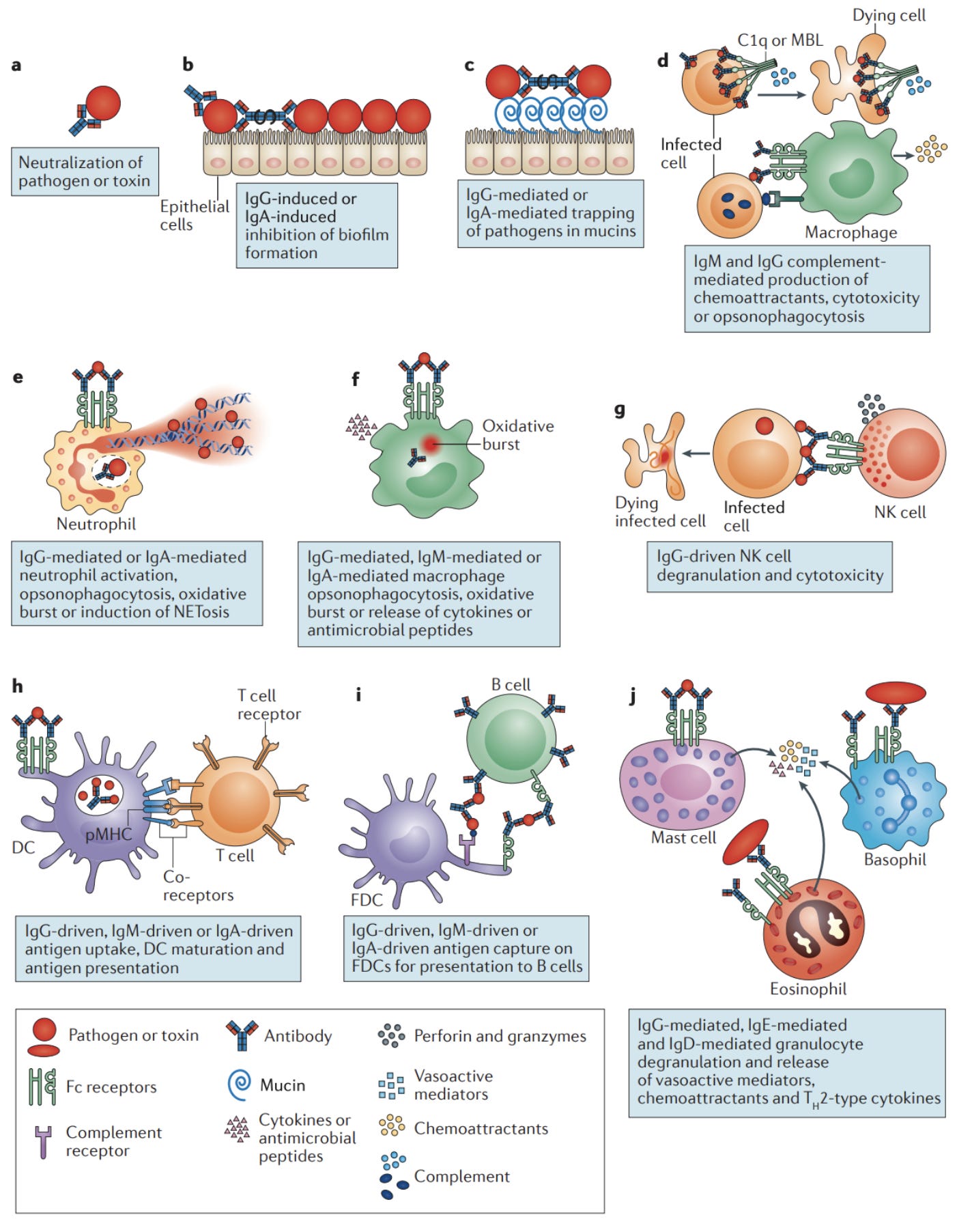
There are many curiosities to the phenomenon of ADE. It’s not entirely clear why some antibody responses seem to be protective and others seem to be pathogenic, but there are many ideas. In fact, in animal models and in cell culture, whether or not an antibody response causes enhanced disease or is responsible for protection is often indistinguishable, hence why they cannot be used to reliably predict whether or not ADE would occur in a human patient. Some have argued that considering how antibodies interact with the pathogen is not sufficient to understand ADE, but rather the effector functions of antibodies are important contributors as well. Antibodies are best known for being neutralizing, but they do many other things as well, depending on the kind of antibody. For example, in addition to antibodies there is a network of proteins throughout the blood called complement. Complement works with antibodies in infection, and is generally considered part of the innate immune system. You can think of them as little bombs in the blood. Complement adheres to the surface of pathogens and eventually forms a structure called the membrane attack complex (MAC) which forms a pore that kills the pathogen (more relevant for bacteria than viruses). In addition, complement proteins can be used to recruit other cells of the immune system and promote inflammation. The complement system is very tightly controlled, and can cause disease by damaging our own cells if control is lacking. The phenomenon of complement-mediated antibody-dependent enhancement (C-ADE) of viral infections has previously been described in the context of HIV. These studies have found that complement activity was found to correlate strongly with viral load and disease progression.
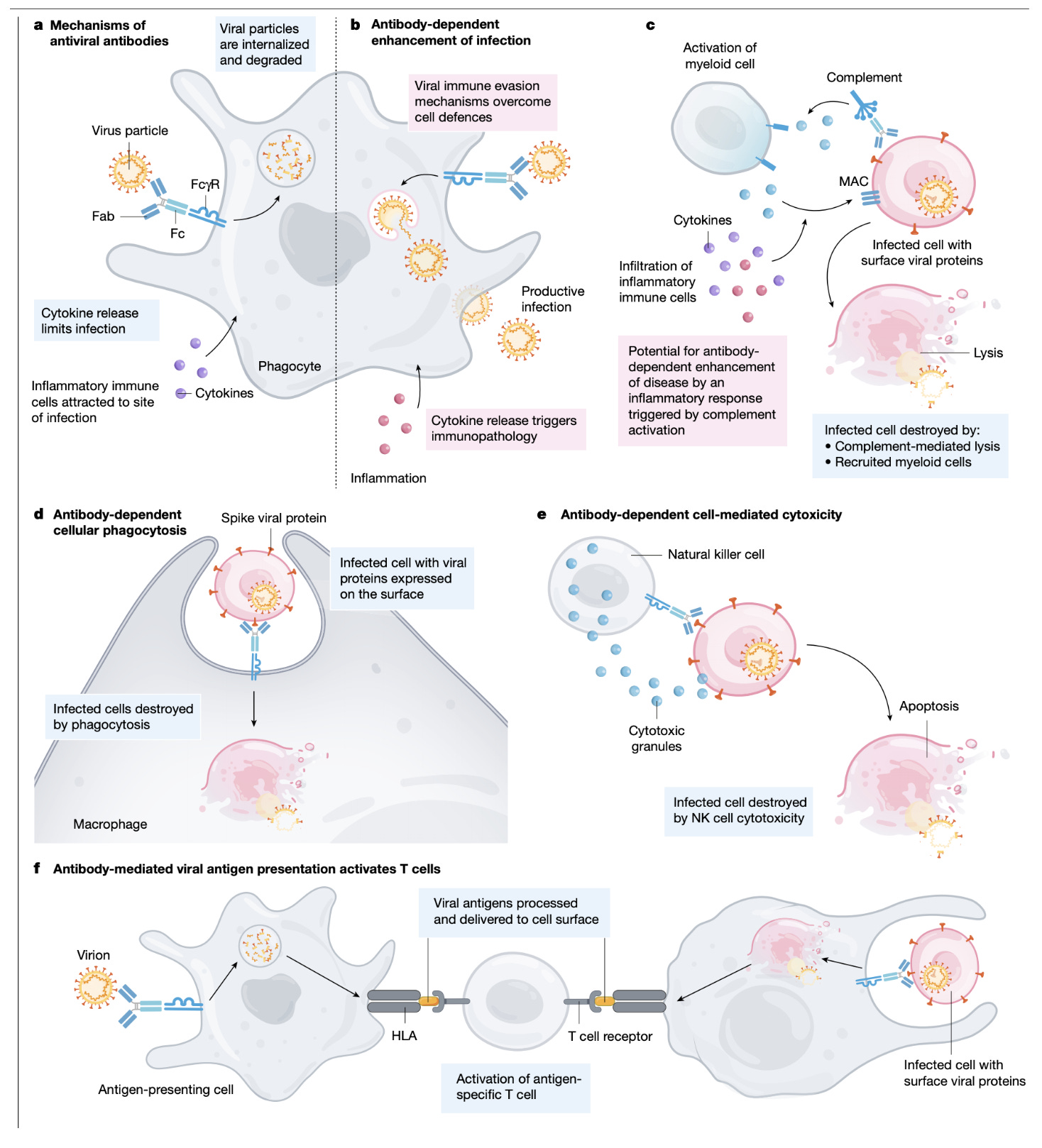

In addition to complement, there are Fc receptor-mediated effector functions of antibodies. These depend a great deal on the specific Fc receptor that the antibodies come into contact with, as this determines the downstream signaling network and response from the cell in question. The FcγR family is the group of Fc receptors that recognize IgG, the major antibody type associated with mature immune responses. Other than FcγRIIb (expressed primarily on B cells), these receptors tend to be activating, promoting the activity of the cells that express them in the context of host defense. In the context of inflammation, activating members of the FcγR family can:
induce respiratory burst from neutrophils (which releases reactive oxygen species, ROS, and reactive nitrogen species, RNS, that while important in host defense, do also cause damage to neighboring cells in a somewhat indiscriminate manner)
mediate antibody-dependent cellular cytotoxicity (wherein antibodies bind to viral proteins still budding off the surface of a cell, triggering an NK cell to kill the infected cell)
promote maturation of dendritic cells (mature dendritic cells go on to license immune responses and need danger signals to do this; FcγRIIb is also expressed on these cells and thus their maturation depends on the relative balance of FcγRIIb compared with activating members of the FcγR family)
Induce the release of cytokines and chemokines that recruit other cells of the immune system to the site of insult
Evidence for the importance of Fc receptors in DHF/DSS comes from studies that demonstrate genetic risk factors for the disease, as some patients express alleles of FcγRIIa with very high affinity for IgG, and also demonstrate heightened affinity of FcγRIIIa for antibodies, which could help to explain the poorly controlled inflammation that is associated with severe disease. Notably, it seems that very high titers of neutralizing antibodies also productively engage FcγR family members and can induce co-activation of multiple members concurrently, but this suppresses pathologic states. ADE has also been noted for Ebola virus, and blocking Fc receptors reduces, but does not eliminate the effect.
Another possible contributor in severe dengue secondary infections is not antibodies at all: it’s T cells. One concept is called original antigenic sin of T cells, and it suggests that T cells against dengue from an initial infection can be primed to respond effectively against that strain, but then respond with low affinity to challenge with a different strain, despite secretion of high levels of pro-inflammatory cytokines like TNF-α. However, the evidence for T cells mediating DHF/DSS is not as robust as that for antibodies, so their contributions are likely less important.
You may begin to wonder how to go about vaccinating against dengue given these challenges. Dengvaxia is a first-generation dengue vaccine (live attenuated, tetravalent to cover all 4 circulating strains), which was to be given to patients who had already experienced a prior dengue infection, and not to those who had not. The issue is that many cases of dengue infection, especially the first infection, are asymptomatic (up to 75% per some estimates), and thus prior to getting the vaccine, serologic evidence was needed to confirm the presence of antibodies. The concept for the vaccine was that it induced neutralizing antibodies against all 4 strains, and maintained them to levels above the critical avidity needed for DHF/DSS to occur. However, the first case of dengue can also cause nonspecific flu-like symptoms, and asking LMICs to serologically test every child from 9 to 16 prior to giving the vaccine was unrealistic. As a result, the warnings were often ignored about not giving dengue to immunologically naive patients with no record of infection, and the vaccine helped to precipitate a number of cases of DHF/DSS. Sanofi, the manufacturer, warned that the vaccine was not to be given to people who were seronegative. Unsurprisingly, vaccine confidence was greatly harmed by this fiasco in the affected regions, especially the Philippines. However, there is perhaps a happy ending here: TV003 demonstrated in a human challenge study that there was NO evidence of DHF/DSS in immunologically naive recipients who were then challenged with dengue virus. So, ADE need not mean a dead end for vaccines.
What about coronaviruses? What about SARS-CoV-2? Wan et al put together a very thorough summary in which they perform a pseudovirus neutralization using MERS-CoV spike protein (this is where a virus is given the gene for a spike protein from a different virus, wherein it becomes known as a pseudovirus as it represents the other virus, and antibody binding is measured). The principles by which it works appear to be very similar to that of dengue and the other viruses for which it is known to occur, which have already been summarized. However, the study in question does rely on pseudovirus neutralization as a surrogate and is done in cell culture, and thus extrapolations to how this affects patient outcome need to be made with much caution. Yang et al performed pseudovirus neutralization assay of a particular SARS-CoV spike protein isolate showed resistance to neutralization and some enhancement of spread in the presence of antibodies for that isolate. However, there was still no clinical evidence of ADE worsening disease. The fact that the effect is so inconsistent in coronaviruses suggests that from the perspective of individual patients and the population, this effect is unlikely to be important, if it occurs at all.
Influenza viruses would be expected to be the poster child for antibody-dependent enhancement. There are many strains and antibodies can be recalled and also be sub-neutralizing- yet influenza surveillance year after year shows no clinical evidence that antibody-dependent enhancement is occurring. Arvin et al summarize the complex evaluation of evidence for this:
Notably, infants benefit from immunization from six months of age, despite their limited capacity to produce affinity-matured, high-avidity antibodies. Overall, widespread annual surveillance of influenza does not reveal ADE of disease, even though cross-reactive strains and vaccine mismatches are common.
So clearly there are still important parts to ADE that need to be figured out.
In summary:
ADE is rare and limited to only a few infections. Even though evidence of ADE can be observed in preclinical models for many pathogens, the cases in which it has demonstrated true clinical evidence are scant.
ADE in vitro does not equate to ADE in vivo.
ADE in an animal model does not, unfortunately, predict ADE in humans.
ADE does not necessarily mean a dead end for vaccine prospects. Dengue, the most famous offender, will soon have a vaccine which does not cause ADE.
ADE does not occur at all levels of antibodies but rather within a specific window, typically as they start to wane. This is explained by a drop in the total avidity of neutralizing antibodies.
Convincing evidence of ADE for COVID-19 has not been demonstrated in nearly a year of this pandemic, despite highly prevalent use of convalescent plasma, indicating it is either very rare, or it doesn’t happen. The conclusion from either outcome is that you should not be concerned about it occurring from a possible COVID-19 vaccine that has passed through clinical trials.
Part III: Non-ADE Mechanisms of Vaccine-induced Disease Enhancement


It’s worth noting however that ADE is not the only way that a vaccine can exacerbate disease. Original antigenic sin of T cells was one proposal for a contributor to severe dengue, and there is also a hypothesis of original antigenic sin with influenza viruses (which is controversial because the evidence for it is inconsistent). But there have been a few instances which have been extremely important for vaccinology where things went very, very bad- and they weren’t because of antibodies.
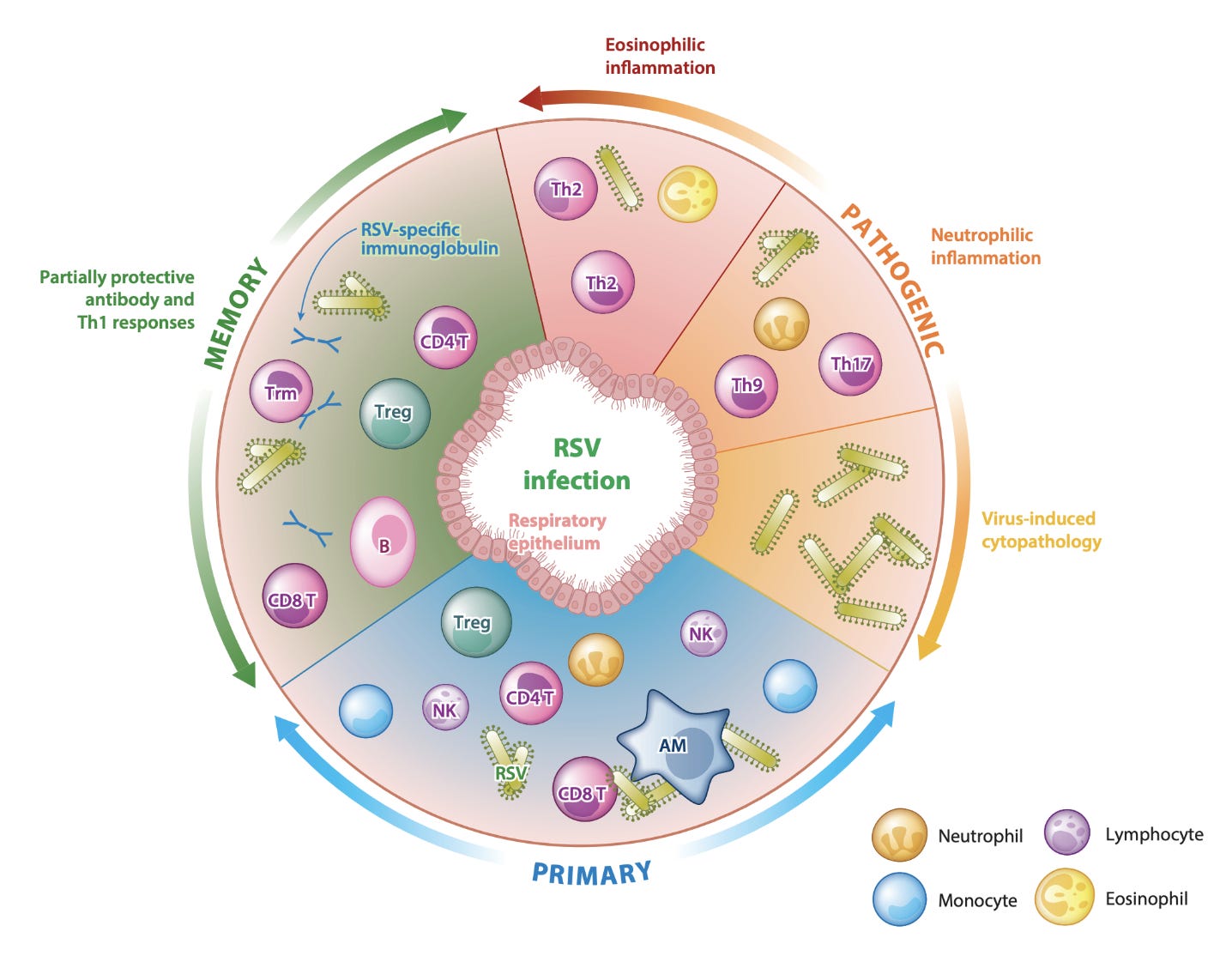
The most famous of these is the respiratory syncytial virus (RSV) vaccine. Feigin and Cherry’s Pediatric Infectious Diseases 8th Edition writes the following regarding RSV:
Respiratory syncytial virus (RSV) is the most important respiratory pathogen of infancy and early childhood and the major cause of hospitalization for bronchiolitis and pneumonia in infants globally. [39,101-103,152,225,228] In the United States, RSV has been estimated to be the leading cause of all hospitalizations for infants, resulting in an estimated 74,000 to 126,000 hospitalizations each year from 1994 to 1996. [175,250] More recent studies suggest that these figures are lower than the current number of infant hospitalizations associated with RSV infections and that the associated annual costs are approximately $2.6 billion. [131,133,145]
So why don’t we have an RSV vaccine if it’s so important for public health? It’s because we haven’t been able to make one successfully. One strategy for making a vaccine is to take the pathogen and inactivate it. This can be done in a number of ways. For instance, you can irradiate it so that its genome is destroyed and it loses the ability to replicate. The most common strategy however is to use heat, or formalin (a dilute solution of formaldehyde). A formalin-inactivated (FI) RSV vaccine was tested in 1965, and the outcome was disastrous. As summarized in Plotkin’s Vaccines 7th Edition:
In the early 1960s, a FI-RSV was prepared and tested in infants and children. This vaccine, designated Lot 100, was administered as two or three intramuscular doses, separated by 1 to 3 months, to infants and children between 2 months and 7 years of age. [24–26,101] Lot 100 not only failed to protect against wild-type (wt) RSV disease, but induced an exaggerated clinical response to wt RSV infection in infants who were RSV naïve before vaccination. Many vaccinees were hospitalized with lower respiratory illness; in one study, the hospitalization rate of vaccinees approached 80% compared with 5% in control vaccinees. [24,25] Tragically, two infants who received Lot 100 died following wt [(wild-type)] RSV infection, one at 14 months and the second at 16 months of age. [24,25] RSV was readily isolated from the lower respiratory tracts of these infants.
Enhanced respiratory disease (ERD) was observed in patients who had received the vaccine, with some of them even having died. Enhanced respiratory disease is what it sounds like- it’s when a prior immune response to a respiratory virus (as far as I am aware it has not been observed with other types of pathogens), whether by vaccine or by prior encounter with that pathogen, worsens disease. At this period in the 1960s, inactivated (killed) vaccines were a very popular concept. They work well for influenza, they are cheap, and they are very easy to make. But they were a terrible idea for the RSV vaccine, as time would tell. Antibodies against RSV from the vaccine turned out not to be neutralizing, and helper T cells primed against the vaccine recruited a cell type called eosinophils. Eosinophils are very important in directing immune responses against helminths (worms), but our understanding of their role in viral infection is incomplete (they are often associated with maladaptive responses, but do appear to have some important direct antiviral roles), and they are important contributors to many allergic diseases (they are also very hard to study because mouse eosinophils are very different from human eosinophils). The evidence for ADE in the case of this RSV vaccine is less convincing than an eosinophil-driven process caused by abnormal T helper cell responses. Most sources report that this was an inappropriate TH2 response (TH2 cells support defenses against helminths and work together with eosinophils and B cells primarily), but Hotez et al argue it is more consistent with a TH17 (TH17 responses are mainly used to deal with extracellular bacteria and work primarily with neutrophils, but some literature does indicate that eosinophils can also play a role) mechanism. There were many ideas for how this may have been induced by the vaccines. A similar phenomenon occurred with the killed measles vaccine (KMV), which was also formalin inactivated, but versions of the RSV vaccine which did not have formalin-inactivated F and G proteins (the major target of the antibody response) also reproduced immunopathology. The proteins themselves seem to be responsible for an inappropriate skewing of the immune response to an abnormal TH2 direction that produces disease. This is consistent with prior studies that suggest that memory T cells themselves have significant plasticity in their choice of effector module, and thus inappropriate skewing may be an inherent feature of the pathogen. Interestingly, administering RSV antigens with a TH1-biasing adjuvant (TH1 cells work with killer T cells and NK cells to support their functions in killing virally infected cells) seems to prevent immunopathology. RSV’s example was used to highlight that vaccines for respiratory viruses merit a special degree of circumspectness in the clinical trial process given the potential for such devastating outcomes.
RSV vaccines were a major reason some people were concerned about the prospect of an inactivated SARS-CoV-2/COVID-19 vaccine. All the advantages about inactivated vaccines- that they are cheap, easy to make, and can often be effective- are still true, but an important study of a vaccine for SARS-CoV (the first one) was especially concerning. Mice were given doubly-inactivated SARS-CoV (the virus was first treated with formalin, and then irradiated with UV light; DIV), and then challenged with SARS-CoV. For some experiments alum was used as an adjuvant (inactivated vaccines generally do not need adjuvants, however), and it seemed to enhance the protection from the DIV. When challenged with isolates of the same type, in young animals, the vaccines seemed to confer protection. However, when challenged with heterologous viruses (distinct isolates), protection did not occur, and in older animals immunopathology occurred with every challenge regardless of whether or not alum were present. Interestingly, alum is a TH2-biasing adjuvant, so TH2 responses are clearly insufficient to explain the immunopathology as alum-adjuvanted vaccines enhanced protection in all groups. Despite this study, there are inactivated COVID-19/SARS-CoV-2 vaccines in Phase III trials, and preliminary findings suggest that at least some of these are well tolerated. Nonetheless, Phase III and Phase IV studies will be needed to confirm their efficacy and continue to monitor for issues with safety. But if there’s any point I hope is clear at this point, it’s that animal and human immune systems are different, so these vaccines should be given a chance- especially in the setting of a pandemic in which they can be rapidly produced in large quantities. The clinical trial process exists to protect us and corners aren’t being cut.
References
Arvin AM, Fink K, Schmid MA, Cathcart A, Spreafico R, Havenar-Daughton C, Lanzavecchia A, Corti D, Virgin HW. 2020. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature. 584(7821):353–363.
Bolles M, Deming D, Long K, Agnihothram S, Whitmore A, Ferris M, Funkhouser W, Gralinski L, Totura A, Heise M, et al. 2011. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J Virol. 85(23):12201–12215.
Bournazos S, Gupta A, Ravetch JV. 2020. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. 20(10):633–643.
Cherry J, Demmler-Harrison GJ, Kaplan SL, Steinbach WJ, Hotez PJ. 2018. Feigin and cherry’s textbook of pediatric infectious diseases: 2-Volume set. 8th ed. Philadelphia, PA: Elsevier - Health Sciences Division.
Clinical Assessment Tourniquet Test. Cdc.gov. https://www.cdc.gov/dengue/training/cme/ccm/page73112.html.
Cohen J. 2019. Critics ‘alarmed by lack of interest’ in studying children put at risk by dengue vaccine. Science. doi:10.1126/science.aaz3853. [accessed 2020 Dec 5]. https://www.sciencemag.org/news/2019/09/critics-alarmed-lack-interest-studying-children-put-risk-dengue-vaccine.
Crotty S. 2018. Do memory CD4 T cells keep their cell-type programming: Plasticity versus fate commitment? Complexities of interpretation due to the heterogeneity of memory CD4 T cells, including T follicular helper cells. Cold Spring Harb Perspect Biol. 10(3). doi:10.1101/cshperspect.a032102. https://cshperspectives.cshlp.org/content/early/2017/04/21/cshperspect.a032102.full.pdf.
Elong Ngono A, Shresta S. 2018. Immune response to dengue and Zika. Annu Rev Immunol. 36(1):279–308.
Halstead SB, Mahalingam S, Marovich MA, Ubol S, Mosser DM. 2010. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect Dis. 10(10):712–722.
Huisman W, Martina BEE, Rimmelzwaan GF, Gruters RA, Osterhaus ADME. 2009. Vaccine-induced enhancement of viral infections. Vaccine. 27(4):505–512.
Keely S, Foster PS. 2015. Stop press: Eosinophils drafted to join the Th17 team. Immunity. 43(1):7–9.
Kirkpatrick BD, Whitehead SS, Pierce KK, Tibery CM, Grier PL, Hynes NA, Larsson CJ, Sabundayo BP, Talaat KR, Janiak A, et al. 2016. The live attenuated dengue vaccine TV003 elicits complete protection against dengue in a human challenge model. Sci Transl Med. 8(330):330ra36.
Kuzmina NA, Younan P, Gilchuk P, Santos RI, Flyak AI, Ilinykh PA, Huang K, Lubaki NM, Ramanathan P, Crowe JE Jr, et al. 2018. Antibody-dependent enhancement of Ebola virus infection by human antibodies isolated from survivors. Cell Rep. 24(7):1802-1815.e5.
Larson HJ, Hartigan-Go K, de Figueiredo A. 2019. Vaccine confidence plummets in the Philippines following dengue vaccine scare: why it matters to pandemic preparedness. Hum Vaccin Immunother. 15(3):625–627.
Lee WS, Wheatley AK, Kent SJ, DeKosky BJ. 2020. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat Microbiol. 5(10):1185–1191.
Lu LL, Suscovich TJ, Fortune SM, Alter G. 2018. Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol. 18(1):46–61.
Openshaw PJM, Chiu C, Culley FJ, Johansson C. 2017. Protective and harmful immunity to RSV infection. Annu Rev Immunol. 35(1):501–532.
Plotkin SA, Orenstein W, Offit DPA, Edwards KM. 2017. Plotkin’s Vaccines. 7th ed. Elsevier.
Prohászka Z, Tóth FD, Bánhegyi D, Füst G. 2006. Complement-mediated antibody-dependent enhancement of viral infections. In: The Complement System. Boston: Kluwer Academic Publishers. p. 249–264.
ROBERT R. RICH MD, THOMAS A. FLEISHER MD, WILLIAM T. SHEARER MD, PhD, HARRY W. SCHROEDER, JR. MD, PhD, ANTHONY J. FREW MD, FRCP, CORNELIA M. WEYAND. 2018. Clinical immunology: Principles and practice. 5th ed. London, England: Elsevier Health Sciences.
Su S, Du L, Jiang S. 2020. Learning from the past: development of safe and effective COVID-19 vaccines. Nat Rev Microbiol. doi:10.1038/s41579-020-00462-y. http://dx.doi.org/10.1038/s41579-020-00462-y.
Thomas S, Redfern JB, Lidbury BA, Mahalingam S. 2006. Antibody-dependent enhancement and vaccine development. Expert Rev Vaccines. 5(4):409–412.
Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, He L, Chen Y, Wu J, Shi Z, et al. 2020. Molecular mechanism for antibody-dependent enhancement of Coronavirus entry. J Virol. 94(5). doi:10.1128/JVI.02015-19. https://jvi.asm.org/content/jvi/94/5/e02015-19.full.pdf.
Weller PF, Spencer LA. 2017. Functions of tissue-resident eosinophils. Nat Rev Immunol. 17(12):746–760.
Wilder-Smith A, Ooi E-E, Horstick O, Wills B. 2019. Dengue. Lancet. 393(10169):350–363.
Xixis KL, Samanta D, Keenaghan M. 2020. Febrile Seizure. In: StatPearls. Treasure Island (FL): StatPearls Publishing.
Yang Z-Y, Werner HC, Kong W-P, Leung K, Traggiai E, Lanzavecchia A, Nabel GJ. 2005. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc Natl Acad Sci U S A. 102(3):797–801.
Yeager A. 2020 Oct 26. More SARS-CoV-2 reinfections reported, but still a rare event. The-scientist.com. [accessed 2020 Dec 4]. https://www.the-scientist.com/news-opinion/more-sars-cov-2-reinfections-reported-but-still-a-rare-event-68089.
Zhang Y, Zeng G, Pan H, Li C, Hu Yaling, Chu K, Han W, Chen Z, Tang R, Yin W, et al. 2020. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18-59 years: a randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect Dis. doi:10.1016/S1473-3099(20)30843-4. http://dx.doi.org/10.1016/S1473-3099(20)30843-4.
Zimmer C, Corum J, Wee S-L. 2020 Jun 10. Coronavirus vaccine tracker. NY Times. [accessed 2020 Dec 5]. https://www.nytimes.com/interactive/2020/science/coronavirus-vaccine-tracker.html.