



Some vaccines are one-and-done. Why aren't they all?
Your immune system is not the whole story.


The current situation with our endemic respiratory viruses (including influenza, RSV1, and SARS-CoV-2- which we have vaccines against) and the resurgence of measles (more or less another respiratory virus) brings a contrast to front of mind: even a single dose of measles vaccine gives durable protection (multiple decades, potentially even life-long) against measles for most people, whereas we regularly need to update our influenza and SARS-CoV-2 vaccines to maintain optimal protection. This raises a question: what does it take to make vaccines work more like those for measles and less like those for flu? I won’t hold you in suspense. The basic factors are:
The type of immune response the vaccine elicits (a bit obvious, but this factor has the most complexity and is also the most modifiable).
How fast the microbe causes disease and spreads, which is related to where the microbe causes infections.
How much variation exists in the microbe with regard to the components targeted by the immune system.
For how these things come together, you’ll have to read the post.
An important note: everything said here is written generally and not specific to individual situations. A vaccine that generally gives antibodies for decades or for life and typically does not require boosters may require a booster in specific individuals, for example, if they have negative antibody titers and will be put at increased risk for contracting the disease (such as with hepatitis B vaccination and work in the healthcare setting). The fact that the vaccine is extremely effective in populations does not negate the need for individuals to get a boosters.
Additionally, this question is obviously huge enough to span many dissertations, so everything said here should be regarded as a highly abridged summary.
Bolded terms are defined in their associated footnotes or in the text surrounding them.
Immune Responses to Vaccination
If you look in an immunology or vaccinology textbook, it will tell you that the large majority of vaccines2 manage their protection through antibodies3. That’s definitely not the only aspect of the immune response important for protection but it is certainly a big one. Antibodies are made by the creatively named antibody-secreting cells (ASCs). These are descendents of B cells (B cells themselves do not secrete antibody) and are split into the plasmablasts and the plasma cells. Plasmablasts are short-lived and help contribute to a huge burst of antibody in the beginning of an immune response, wherein they may differentiate further into plasma cells or die (most will die). The antibodies they produce however will persist depending on their half-life (about 3-4 weeks for most IgG, 4-7 days for IgA, etc.). Some plasmablasts and B cells may differentiate into plasma cells. Plasma cells are extremely specialized for the secretion of antibody, capable of making huge amounts of it for as long as they live.

Therein lies the challenge: plasma cells can be long-lived (surviving for decades, even your entire lifetime) or short-lived. Hence, the holy grail of vaccinology would be vaccines that reliably induce lots of long-lived plasma cells (LLPCs). But… no one is exactly sure what it takes to get LLPCs,4 though we do know quite a bit:
The longevity of plasma cells depends in part on their ability to access survival niches located within the bone marrow and the mucosae. It is likely that most LLPCs are generated within the survival niches themselves, mainly after migrating to them at the plasmablast stage.
The valency5 of the immunogen6 has a major role, with higher immunogen valency favoring plasma cell generation. Vaccines with higher valency immunogens (virus-like particles and live attenuated viruses for example) tend to give more durable antibody responses.
The physical distance between the epitopes7 on multivalent immunogen displays also plays a role, with smaller distances favoring stronger antibody responses (which is thought to be closely related to the effect of valency).
The size of the immunogen is important for ensuring it reaches the correct regions of the lymph node to stimulate the most effective antibody responses (if too large it will simply sit at the injection site and if too small it will rapidly by cleared in the bloodstream and fail to give much of a response; ideally immunogens get maximally taken up into the lymph nodes to orchestrate responses).
Getting T cells to help B cells helps tremendously with generating LLPCs (but is not essential).
Certain metabolic requirements for the maintenance of LLPCs are now known (reviewed more here). How these could be used to maximize the effectiveness of vaccination is an area of active research.
Vaccines and infections9 differ greatly in their ability to induce LLPCs. Influenza vaccines for example seem to struggle to generate plasma cells lasting longer than about 1 year. Analogous studies for the infection are lacking. It is commonly reported that influenza infection may generate lifelong humoral immunity against matched strains based on the finding that individuals who lived through the 1918 pandemic had antibodies that recognized that virus well into their 90s; however, this ignores the fact that these individuals were reinfected with influenza throughout their lifetimes, including by viruses closely related to the 1918 strain, which could account for their retained ability to recognize the 1918 virus (in fact this is probably the case given the extremely degree of mutation observed in the antibodies). Measles vaccination or infection reliably generates antibody responses lasting many decades, if not life, as does orthopoxvirus vaccination and infection.
Still, LLPCs are all well and good but there’s a very serious weakness in the protection they provide: LLPCs are not capable of undergoing affinity10 maturation. Affinity maturation refers to a process in which B cells mutate the DNA encoding the B cell receptor and test it against the immunogen, seeking to get help from a T cell by presenting that immunogen to it. The B cells with the highest affinity receptors will bind the immunogen most tightly, and thereby be able to get the most help from T cells and become antibody-secreting or memory11 B cells (Bmem). B cells that mutate to a receptor that is less effective or cannot bind the immunogen will not be able to receive help and will die. This occurs with re-exposures to the immunogen as well, and so the affinity of antibodies can rise greatly from primary to secondary immune to tertiary etc. responses:

This process allows us to develop antibodies that bind antigens more tightly and also more broadly, such as if new variants emerge that can escape the old antibodies. With long-lived plasma cells, the antibody they secrete could become useless with the emergence of a new variant (if the old one also disappears), and yet it will persist because the plasma cell persists, leaving us susceptible to infection. Memory B cells however, address this gap by retaining the ability to rapidly differentiate into plasma cells, undergo affinity maturation, and persisting long after the threat has been cleared. Thus, when it comes to B cell immunity, we have “two walls” as this review conceptualized:
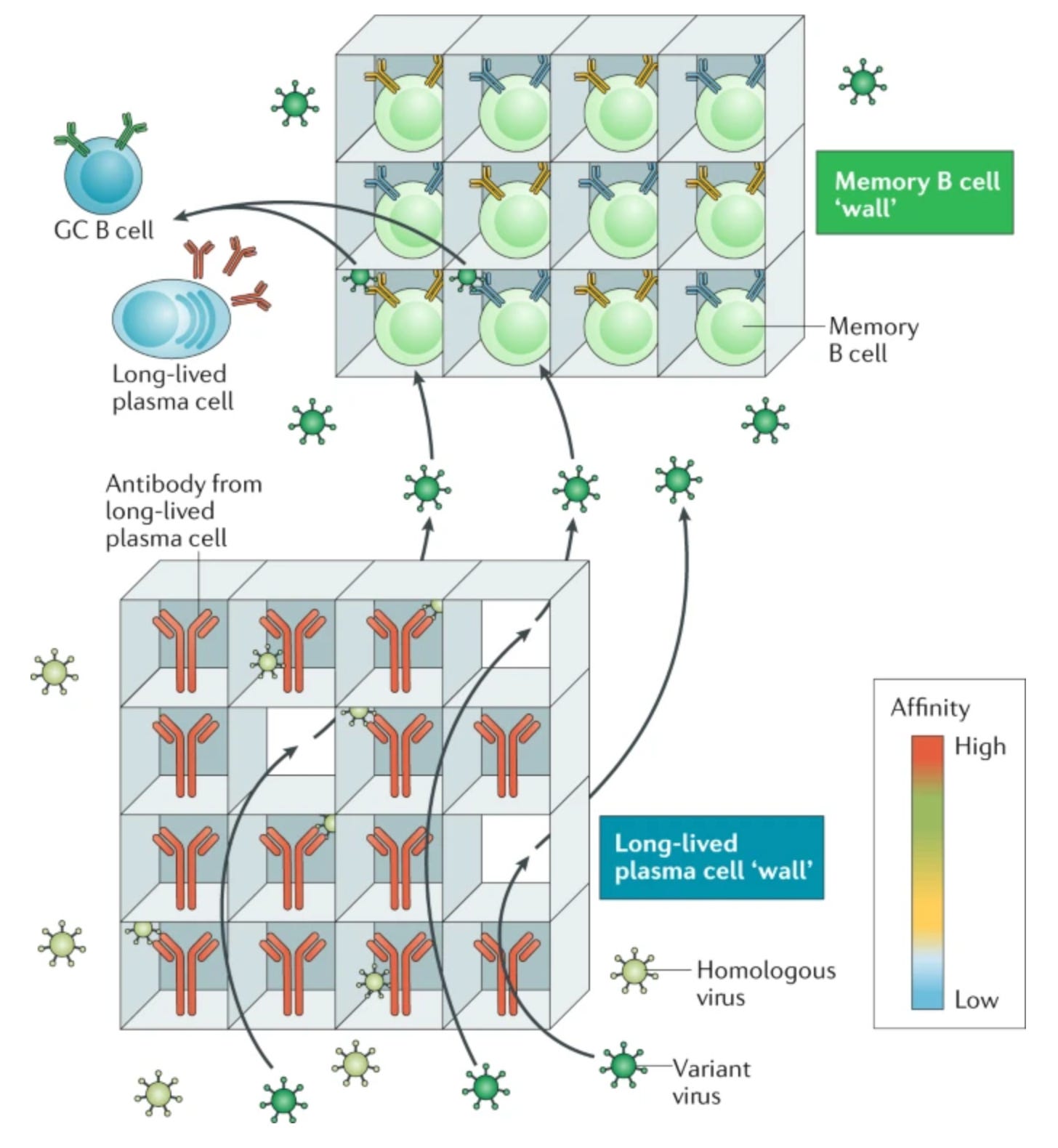
There is a lot to be said about how antibodies work to generate protection from vaccination, as there are many misconceptions there (in particular, there is a broad ignorance about the roles of antibodies beyond neutralization12, and the diversity of mechanisms by which neutralization can be achieved). For the purposes of this post, I will say that often the ability of an antibody to neutralize some target antigen is a correlate of protection (CoP)13 for vaccination, particularly for symptomatic infections.
Because antibodies are the effectors14 of neutralization, they are responsible for sterilizing immunity15. However, this does not mean that antibodies are the only effectors of the immune response relevant to vaccination, because prevention of infections is not the only outcome relevant to the protection of vaccination. In fact, there is no vaccine that has been shown to prevent infection completely (in large part because the technology to verify this does not exist)- but this has not stopped vaccines from having extraordinary effects on demolishing the incidence of vaccine-preventable diseases. Still, we should recall the dragon in Sagan’s garage and ask ourselves whether there is a difference between an untransmissible, asymptomatic infection and no infection at all.
T cells
In addition to the B cell arm of the immune system, we have the T cell arm. T cells are broadly split into helper and killer T cells, which are easily distinguished by their expression of the proteins CD4 and CD8, respectively16. Essentially all adaptive immunity17 depends on helper T cells (the major exception to this is responses to polysaccharide immunogens, which B cells can manage on their own, but these responses tend to be much less durable and smaller than those involving T cells). CD4 T cells are undeniably indispensable for protection. We can deplete people’s B cells18 (as is done in some forms of cancer or the management of autoimmune disease) and they retain the ability to recover from infections (including COVID-19). If even just the CD4 T cells are allowed to be depleted (as happens with uncontrolled HIV infection), the result is the onset of opportunistic infections which are, eventually, invariably fatal19.
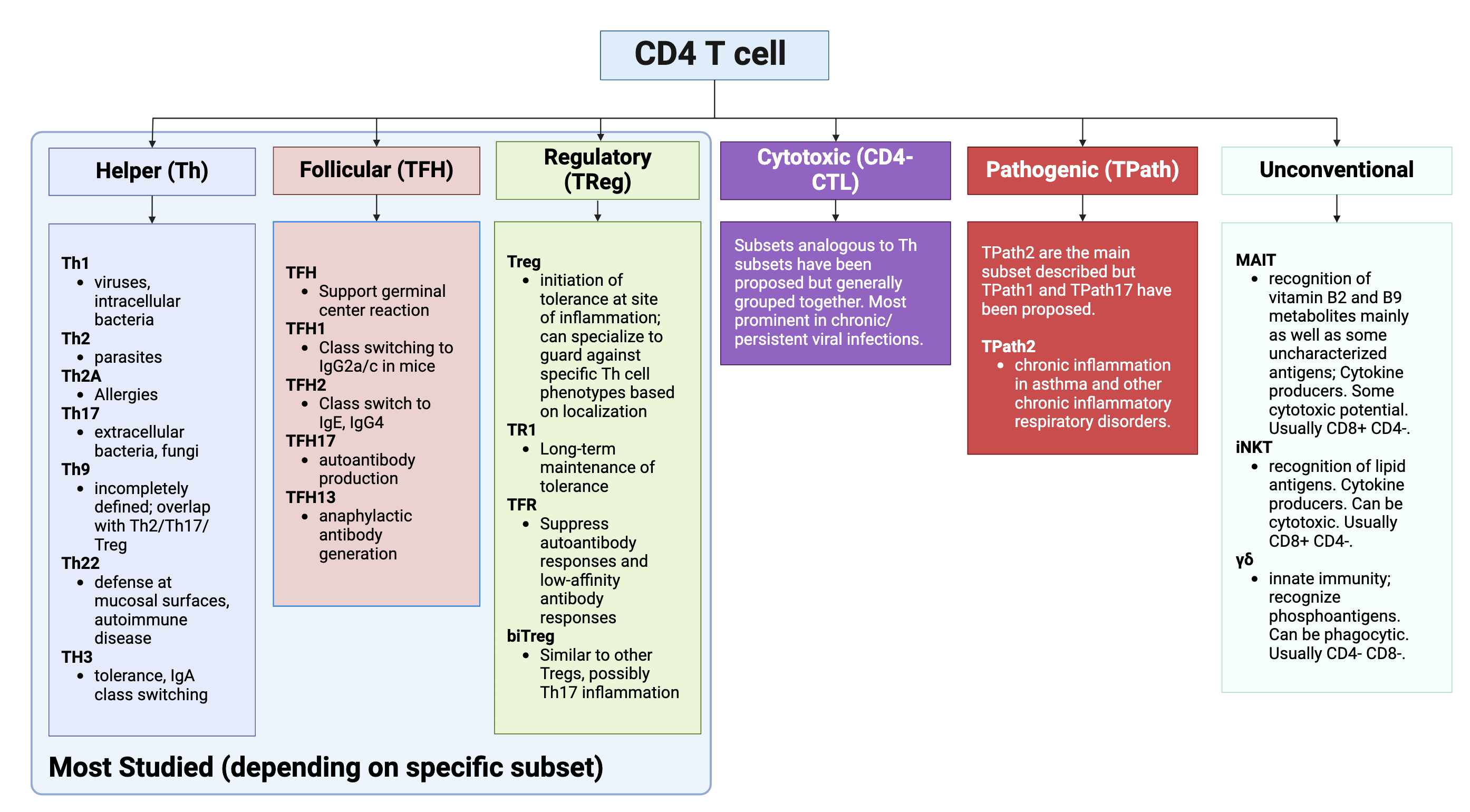
The major role of CD4 T cells is as helpers to polarize the immune response in the right direction to protect the host. There is a distinct type of helper T cell for each type of immune response, and the diversity of CD4 T cells is staggering, as described below (this is for illustrative purposes only, and believe it or not this isn’t even all of them- you should not feel pressured to learn every single type of CD4 T cell):
Vaccines vary greatly in their ability to stimulate TFH cells, which support antibody responses20. Different adjuvants can support the development of TFH cells to different degrees. Similarly, vaccines differ in their ability to induce different T helper cells. For example, alum is one of the most common vaccine adjuvants and it supports Th2 responses, which are effective against toxins such as those made by diphtheria, tetanus, and pertussis (though that’s not their evolved purpose). The unmodified nucleic acids present in live attenuated viruses enable Th1 and killer T cell responses that are evolved specifically for that purpose (in effect, serving as adjuvants).
The scope of CD8 T cell responses is more limited. Classically, the function of CD8 T cells is to eliminate virally infected cells and tumors from the body by killing them. However, CD8 T cells have also been shown to contribute to defense against intracellular bacteria, some fungi, and some intracellular parasites as well. Despite this, deficiency of CD8 T cells alone while other parts of the immune system are intact, while a true immunodeficiency, is not especially severe (and is exceptionally rare) compared to the effect of lacking CD4 T cells21. Subsets of the CD8 T cells largely mirror those of CD4 T cells, with regulatory CD8 T cells, follicular CD8 T cells, and helper-like CD8 T cells that produce cytokines all being described.
In addition to the immune module elicited, another factor to consider is the type of memory T cell you get. These can be classified more granularly, but in general we only need to consider 3 types:
Effector memory (TEM): Memory cells specialized for entering inflamed tissues, capable of circulating throughout the entire body. TEMs are capable of maturing rapidly into effector T cells that deal directly with immunological threats, but limited capacity to divide and proliferate, which is thought to limit their effectiveness in controlling infections.
Central Memory (TCM): Central memory T cells circulate between the blood and the lymph nodes like naive 22 T cells, but have extremely high sensitivity to the antigen they are specific to. Upon recognizing that antigen, they can rapidly differentiate into effector T cells which can deal with the threat directly. TCMs are associated with more effective control of infections than effector memory cells, but context is important.
Tissue-Resident Memory (TRM): These are induced at the site of infection or vaccination and retained, allowing them to respond more rapidly to threats than other memory T cell subsets because they don’t need to migrate to the site of infection. The conditions for eliciting them varies across body sites and it is thought that in some cases they are replenished from effector memory cells.
It is thought that the balance between TCM and TEM cells is dictated by the persistence of antigen, with shorter duration of antigen persistence favoring TCM induction and persistence favoring TEM induction. On this basis, it has been suggested that replicating vaccines induce better protection than non-replicating vaccines because they result in greater antigen persistence, favoring more TEM cells. Despite this, the yellow fever vaccine, which is a replicating vaccine, has been shown to induce a population of T cells known as stem cell memory T cells (TSCMs, which are similar to TCMs), which is thought to account (in part) for its exceptional durability of protection (a single dose generally confers lifelong protection). The Pfizer/BioNTech COVID-19 mRNA vaccine has also been shown to elicit these types of T cells as well.
Despite the obviously central role T cells play in the immune response, and therefore the protection elicited by vaccines, the details regarding how this is accomplished for basically all vaccines is not well understood. Classically, the role of T cells in vaccines has been appreciated essentially only for their ability to enhance antibody responses.
Understanding how vaccines induce protection and their limitations is critical to addressing the question of how to get more effective, durable vaccines because of all the factors that determine that outcome, this one is probably the most modifiable.
Microbe Replication: Everything is Kinetics and Kinetics is Everything
Within the field of vaccinology, it is commonly said that mucosal infections (those that occur at the mucosal surfaces such as the respiratory tract, gastrointestinal tract, and urogenital tract- all the slimy things that are in direct contact with the outside world) are much harder to control with vaccination than those that spread through the blood and cause systemic infections. It is true that immunity is a somewhat compartmentalized, and the mucosal immune system has different adaptations to deal with infection than the systemic immune system, as inflammation23 at the mucosal surfaces (such as the lung) can be especially costly in terms of its harms to the host. You can see it in the trends in durable immunity to various microbes:

In the same vein, it has been reported in the past that the durability of antibody responses to pathogens24 that infect the mucosae is fundamentally poorer:
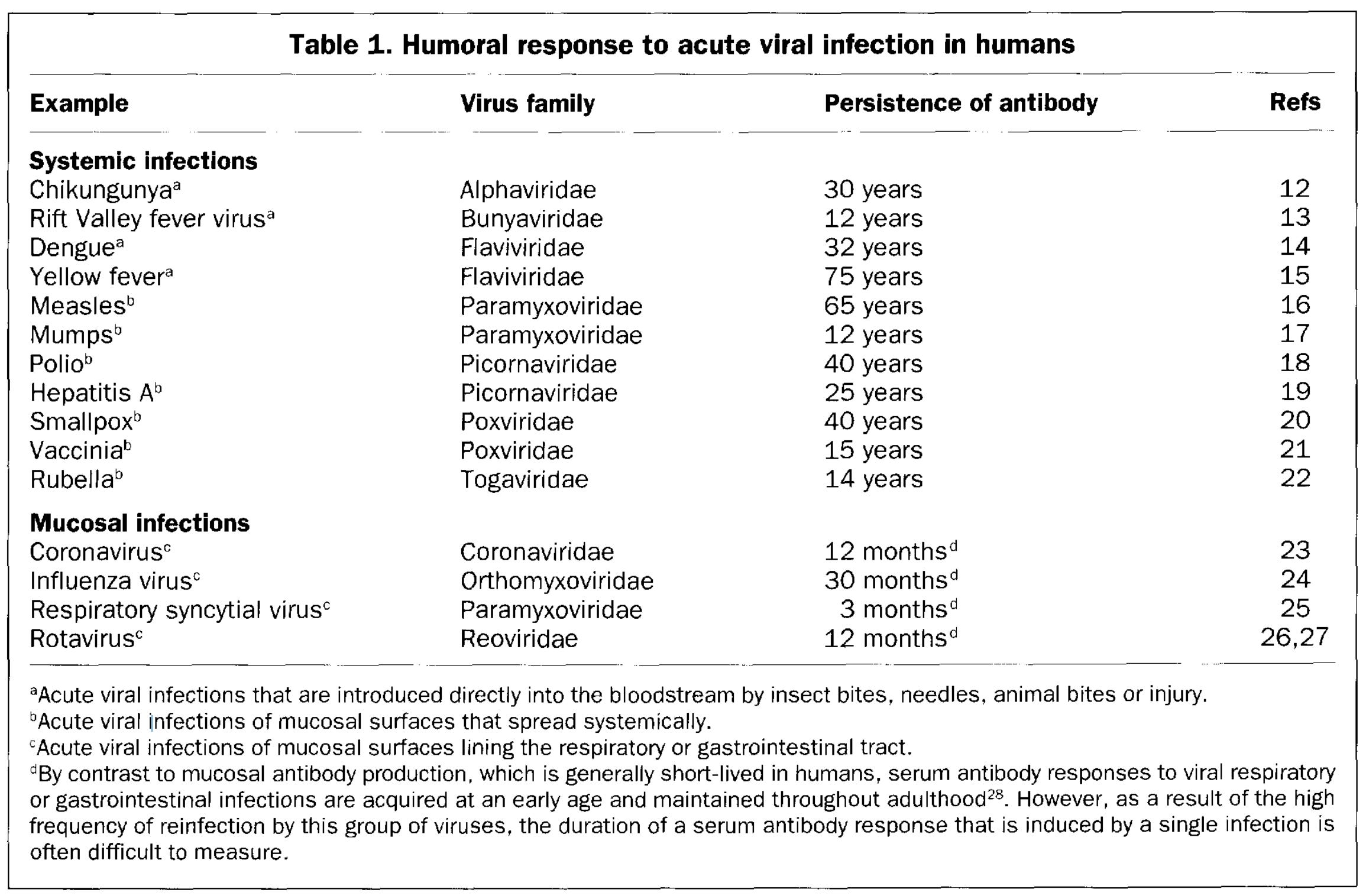
The problem with these metrics however is they are looking specifically at antibodies in the blood, leading to the perception that mucosal infections do not establish long-lived protection. This is probably not the case (obviously it will vary from microbe to microbe), but it wouldn’t be unusual for these not to be captured in the blood given current models for how such protection works25:

Why then, do we see this trend with mucosal vs systemic infections? Every infection you experience is a race between your immune system and the pathogen trying to use your body to meet its evolutionary imperatives. By virtue of their different metabolic needs, in general, microbes replicate much faster than the cells of our immune system (and immune cells replicate much faster than most of the cells in our body). For example, bacteria can increase their population tenfold in about 66 minutes, and viruses can replicate at comparable rates. Estimates vary widely and by subset of cells, but it has been reported that it takes about 24 hours for memory T cells to increase their concentration tenfold; for naive T cells this degree of replication may require as long as 7640 days. In any straightforward race, the immune system will lose. The caveat here is that these microbe replication speeds are attained under ideal conditions- something that the immune system rapidly works to remove from the equation. For example, many immune cells have markedly enhanced activity at temperatures a bit above body temperatures whereas many microbes have reduced replication at these- hence the value of fever. The cells of the body are also evolved to resist infection. For example, virtually every cell in the body has the ability to make type I interferon in response to a viral infection, serving to interrupt the metabolic processes that support viral replication, activate the cells of the immune system, and mark the infected cell as being infected so it can be cleared. The trouble is… virtually every single virus that causes disease has ways to subvert the effects of interferon (or else they would not be able to infect and cause disease). So, the microbes are armed too. In some sense, the fundamental point of vaccination is to give your immune system as much of an advantage as it can have against these pathogens in this race- because they aren’t playing fair.
Thus, the mucosal vs. systemic infection issue might be missing a key point:

Admittedly, the review that table is sourced from is a bit dated (reflected in some of its comments about the immunology- for example, there is no mention of long-lived plasma cells) but the basic point is true. It is so much harder to prevent an infection that is fast than one that is slow. The pace of infections is roughly reflected in whether or not post-exposure vaccination to prevent disease is possible (which it is for measles, smallpox, mumps, hepatitis A, hepatitis B, rabies, anthrax, and varicella). It’s worth noting that in human challenge studies where the same strain of influenza is used for infection twice, one year apart, a majority of people still develop influenza illness a second time a year later.
One challenge is that if primed by a systemic immune response26 (such as that which follows an intramuscular injection), it is thought that even though the immune system has demonstrated an ability to mount recall responses in as soon as 2-7 days following infections, is is thought that the antigens in a mucosal infection may not penetrate into the systemic compartment (e.g. the bloodstream) and thus the recall response may be unable to outrun the pathogenesis of the organism. This is specifically highlighted as a problem with acellular pertussis vaccines, which is working on being corrected with novel pertussis vaccines.
Still, the fast infections aren’t going to stop being fast, so what do we do about them? One option is boosting27. Rather than rely on recall responses to keep us from getting seriously ill (which they should generally do), we preventively get additional vaccine doses to better prevent infections or accelerate their control. This is the strategy for influenza, COVID-19, pertussis, etc. Its success varies, and is often hampered by the need for high uptake of the vaccine to make a difference. Another option is to target vaccines to the site of infection with the intent of inducing tissue-resident memory cells that will mount more rapid recall responses. The success of this approach is kind of a mixed bag. For polio, the oral polio vaccine annihilates transmission chains in a way that the inactivated vaccine simply cannot (though the inactivated vaccine is safer and does reduce spread to a lesser extent). For influenza vaccination, the nasal vaccine doesn’t seem to do any better (and possibly does worse) than the injected one in adults (the reasons behind this are complex) but might have an advantage in young kids.
Escaping Immune Responses
The ability of a pathogen to cause an infection in an immunologically experienced host requires some degree of evasion from the host’s immune system. Antibodies are often spoken about as a first-line defense against infection (which is true if you disregard the innate immune system28), with the intention being that the microbe will encounter the antibodies and then fail to initiate infection as it is cleared by the immune system or at least have its replication restrained enough that the immune system can rapidly abort the infection. The trouble is that, assuming antibody levels at the site of infection are have high enough titers to do this, they still have to be able to bind the pathogen, which creates a selection pressure: the variants that escape this first line of defense will be able to cause infection (though not necessarily disease). As pathogens replicate, they make errors and give rise to imperfect copies that differ from one another, and some of these might be able to effectively evade variants. In other words, these kinds of occurrences push us backwards to a state closer to immunological naivete. This phenomenon is known as antigenic drift.
Sometimes, our own evolved defenses against these pathogens can backfire on us. For example, humans have a family of enzymes known as APOBEC3s (apolipoprotein B editing complex 3) which mutate the genomes of viruses that infect cells (where the mutations occur depends on the specific enzyme, but in general they replace C’s with U’s). Another family of enzymes known as ADARs (Adenosine deaminase acting on RNA) works in a similar manner. These enzymes induce missense and nonsense mutations29 which reduce the efficiency of virus replication, and therefore onward transmission. In the case of HIV, they can make its ~1 in 10,000 replication error rate go up to 1 in 70. Unfortunately, on occasion viruses can persist despite these mutations, and thus we see mutational jumps. This has also been seen in mpox, SARS-CoV-2, and more.
For infections that are of short duration though, the ability to jump to a variant with strong evasion of pre-existing antibodies is pretty limited. This is why the large jumps in the evolution of SARS-CoV-2 (known as saltations) likely come from chronically infected cases. Another source of rapid evolution is recombination, wherein two (or more, potentially) different variants infect the same cell and exchange genome segments. In the case of bacteria, horizontal gene transfer may also account for significant declines in the ability of pre-existing immunity to protect. Another potential factor in some cases is loss of the relevant immunogen. For example, one of the major antigens in the acellular pertussis vaccine is the protein pertactin, found on the bacteria’s surface. Regions where the acellular vaccine is used have seen a rise in pertactin-deficient strains of Bordetella pertussis, enabling infections to occur (indeed, infection with Bordetella pertussis appears to be much more common than previously appreciated, as T cell responses against the bacteria are very broad even against antigens not present in the vaccines, indicating undetected infections). Despite this, evidence suggests that the vaccines retain significant protection against pertussis disease even in the setting of widespread pertactin deficiency, consistent with the central role of pertussis toxin in the disease.
Because T cells can only act once an infection has already occurred and because of the pace of immune responses relative to transmission, evolution to escape them has been much rarer in the context of SARS-CoV-2. However, (some) escape from killer T cells has been observed in chronic infections. This is also seen in other chronic infections, such as hepatitis C virus (although the chronic nature of the infection there also results in exhaustion of the the T cells). Here the speed of infection plays a key role too: influenza infection is so fast that it appears to spread before there is any level of meaningful selection for escape variants at the individual level, but not so fast that there is none at the population level (accounting for why flu seems to mutate so quickly to escape as the season progresses).
Pathogens do face constraints on their ability to escape pre-existing immunity, at least in theory. For example, escaping an immune response may come at the cost of losing the ability to productively interact with the receptor that enables infection. This appears to be the case with measles, and thus the observed inability of the virus to escape pre-existing immunity. However, pathogens do have an unpleasant trick: they have the ability to change which receptors they use to cause infection. For example, HIV-1 (which is responsible for most cases of HIV) infects CD4 T cells, but they need to express a coreceptor to actually enable the infection. When contracted by the mucosal route (such as through exposure to bodily fluids), as occurs in most cases of transmission, this is typically a protein called CCR5, so most HIV infections begin with so-called R5 viruses (also known as M-tropic viruses because they infect macrophages). Some people naturally have a deletion in CCR5 called ∆32, which renders them resistant to HIV infection (in fact, a few individuals with HIV have been cured by bone marrow transplantation from donors with two copies of this mutation). However, HIV can mutate to rely on CXCR4 (such viruses are known as X4 viruses, aka T-tropic, because they infect T cells, though R5 viruses also infect T cells) for entry, which would render these susceptible. A subset of HIV viruses can use both receptors (dual-tropic, grouped into dual-R and dual-X based on which receptor is preferred).
The Coronavirus in the Room: What this means for SARS-CoV-2 Vaccines
These factors suggest that a vaccine offering durable protection against SARS-CoV-2 infection without the need for boosters will be, to put it mildly, an extreme challenge. To start with, SARS-CoV-2 itself does not prevent symptomatic reinfections indefinitely, particularly with antigenic saltation events like the emergence of Omicron. It is not impossible to get vaccines that outperform infections in terms of their protection (in fact, there’s evidence that this may in fact be the case for mRNA vaccines when it comes to severe infections with SARS-CoV-2), but this helps to establish a baseline for the degree to which that is feasible- the vaccines that give durable protection tend to be against infections that also give durable protection.
SARS-CoV-2 gave a painful education on the role of replication speed in the ability to cause disease:

The delta variant marked a major turning point in the ability of vaccines to prevent symptomatic infection, prompting considerations for the value and necessity of booster doses30. Virus titers from delta peak at a much higher level and earlier than the ancestral variant, fundamentally giving the immune system less time to mount a response to suppress both transmission and the development of disease. The delta variant also had some degree of antibody evasion but it was significantly less than that of the beta variant in most studies. The change to the replication speed of Delta is very much underappreciated in discussing the effect on vaccine effectiveness. When Omicron emerged, the virus demonstrated an unprecedented level of antibody evasion, but also showed even more rapid replication kinetics in the upper respiratory tract than Delta. These things together have made the durable suppression of milder infections and transmission a huge challenge.
SARS-CoV-2 infection has been suggested to elicit LLPCs in the bone marrow, but they appear to be significantly less common than those observed for tetanus toxoid and account for a minority of the overall antibody response. Cells that match the description of LLPCs have also been reported after vaccination with the Pfizer mRNA vaccine, but some have argued that these may represent cells induced by infection which went undetected. Nonetheless, the antibody titers that matter most when it comes to infections are those in the upper respiratory tract, the site of infection by SARS-CoV-2. Unfortunately, the picture there looks fairly pessimistic. Even though infection does elicit sIgA antibodies that are potent neutralizers (and in some cases very high levels), they appear to be fairly short-lived (although they do not drop all the way to 0, indicating the retention of plasma cells). However, there is cause for optimism:

Vaccination in those with infection experience restores upper respiratory antibody levels, even with vaccines given intramuscularly (in this study they received Oxford/AZ or mRNA). This suggests that there might be a version of events where we can boost our way to suppressing infections. This also suggests that the gains from a mucosal vaccine, in a world where so many people have already been infected with SARS-CoV-2, might not be as substantial as we hope. Indeed, T cells induced by vaccination in those who have previously been infected traffic to the upper airway. There is, however, another challenge.
The gains made by SARS-CoV-2 during this pandemic are also attributable in part to its ability to evade pre-existing antibodies. At this point, the receptor-binding domain (RBD) of the spike protein is undergoing convergent evolution at a fairly rapid pace. The natural tendency of the immune system is to focus on the conserved features of any given immunogen relative to its prior experience. Hence, when a spike protein with a very different RBD but with other parts of the spike protein well conserved, the immune response will focus on the conserved features over the novel ones. This is sometimes known by the terms original antigenic sin, imprinting, antigenic seniority, or primary addiction31:

The advantage of such an approach is obvious: a major key to protection against infections is to respond as quickly as possible, and memory cells can be recruited much more rapidly to deal with threats. This also helps you to evolve antibodies with very high breadth (see also this preprint)- the ability to cover many variants. In fact, an antibody (SC27) from a person receiving the Pfizer mRNA vaccine and having a breakthrough infection demonstrates the ability to neutralize every variant of SARS-CoV-2 tested, several SARS-like viruses, including SARS-CoV-1, with a binding affinity beyond that which was even though to be theoretically possible and demonstrates potent protection in an animal model. However, because the RBD is so variable across variants, the immune response on boosting focuses on less potent epitopes. Here, however, there is also a potential solution: excluding the more conserved epitopes:

The neutralizing antibody response to BA.5 RBD-only vaccines in mice after priming with whole ancestral shows a marked advantage over the whole RBD spike (22-fold over whole BA.5 spike, 50 fold over ancestral spike against BA.5; 23-fold over ancestral and 15-fold over BA.5 whole spike for XBB.1.5). The exciting part about this is that there is actually a vaccine in phase 3 trials which includes just RBD and NTD: mRNA-1283. Even at one-tenth the dose (in those who haven’t been vaccinated or infected), the vaccine gives comparable neutralization to Moderna’s whole spike mRNA-1273, but with less reactogenicity32:

Data on mRNA-1283’s immunogenicity as a booster to those with immunological experience, however, are not yet available (to the best of my knowledge), but Moderna does plan to study it in this capacity. mRNA of course does not have to be the only platform. For example, Corbevax is a patent-free protein-based vaccine containing the RBD of the spike protein adjuvanted with alum and CpG-1018 which the WHO has approved; the FDA however has not approved it.
There is a lot more that can be said about the potential avenues that can be taken to address the shortcomings of our COVID-19 vaccines (and influenza for that matter), but they are perhaps best reserved for a future post. Nonetheless, the fundamental characteristics of SARS-CoV-2 are such that controlling the spread will not be possible without regular boosters because the pace of infection and antigenic drift are too rapid.
Although this post is about the need for boosters, we currently have no real guidelines to inform the use of boosters for RSV vaccines. The data we have on RSV vaccines for older adults show that the vaccines retain their protective effect through a second season, and, at least for GSK’s vaccine, a second dose does not improve upon protection. This suggests that regular boosters of RSV vaccines may not be required for the control of RSV-associated morbidity and mortality, though it does not necessarily mean that boosters will never be needed. The immune system of older adults is infamously subject to the phenomenon of immunosenescence, wherein with advancing age, it becomes more difficult to mount competent, durable immune responses. This coupled with the burden of comorbid conditions that come with advancing age could mean additional doses may still be needed in the future at some point, but the early data suggest probably not as an annual occurrence in the way we have for flu or SARS-CoV-2.
There is similarly no guidance regarding the use of additional doses in pregnancy; In general, I would expect these to follow the trends for other routine pregnancy immunizations like those for pertussis, tetanus, influenza, and SARS-CoV-2, wherein with each pregnancy a dose is recommended to maximize the amount of antibody transferred (and therefore protection) across the placenta to protect the infant, but there are currently no data to inform these recommendations. Data on immunogenicity and safety are urgently needed.
A potential caveat with all of this is that RSV’s F protein is stable with regard to antigenic evolution, which, on its face inspires optimism. However, at least part of this is likely because of the fact that structurally, RSV’s F protein is metastable and hides the most potent neutralizing epitopes from antibodies in the prefusion conformation and RSV infection appears to be limited in its ability to boost the PreF titers. Through the use of vaccines that stabilize F in its prefusion conformation, antibody titers far beyond what has ever been achievable through infection are possible, which may mean stronger selection for escape variants. For this reason, I would argue that it is key to maintain genomic and antigenic surveillance of this virus in the coming years to assess the risk of antibody escape. Notably, RSV monoclonal antibodies have historically been shown to select for escape mutants and some have even failed because of the emergence of resistant isolates (although Nirsevimab has been reported to be quite robust). It is considerably more difficult to evolve escape to all of the antibodies elicited by a vaccine (as they target multiple parts of the F protein at the same time) than to a monoclonal antibody targeting just one epitope. At the same time, it is hard to gauge how much the F protein can change while preserving the ability of the virus to infect cells because the receptor for RSV is not well-defined (we know multiple receptors used by RSV, but how much each one contributes to infection in the body is less clear), as has been done for other viruses such as measles and SARS-CoV-2.
It may seem a bit silly to define vaccines, but for some reason this has caused a bit of a kerfuffle among some members of the general public so…
Vaccine: Any preparation of an immunogen that elicits immunological memory to protect from the negative effects of that immunogen, related immunogens, or substances inherently associated with that immunogen. These include protection from infection, colonization, disease, severe disease, death, or transmission (when applicable). It is possible for a vaccine to offer protection against only some of these or offer much better protection against some of these outcomes than others.
Under this definition, allergen immunotherapy is also considered a vaccine. In contrast to vaccination against infectious diseases or cancer, allergen immunotherapy aims to tolerize the body to the immunogen; however, the mechanisms involved overlap with those of vaccination against infectious diseases (for example, elicitation of IgG antibodies to compete with IgE antibodies for binding) but also have unique aspects (for example, administration at higher doses and at much higher frequency to elicit regulatory T cells that will suppress allergic responses against the allergen).
I would not consider a preparation lacking an immunogen to be a vaccine even if the preparation induces immunological memory. For example, a cocktail of adjuvants was recently shown in mice to enabled broad protection against a variety of multidrug-resistant microbes via trained immunity. For the moment I would regard this as being an immunomodulator, rather than a vaccine (though the authors do call it a vaccine). Admittedly, this might be prejudicial on my part as I am in effect segregating the immunological memory demonstrated by lymphocytes from the trained immunity exhibited by the components of the innate immune system.
There are cases where I think it becomes fuzzier. For example, antibodies can be used to target antigens on the surface of tumors and used as cancer immunotherapy. These can furthermore be engineered to engage with the protein CD32A on antigen-presenting cells to enable the development of immunological memory against tumor antigens, in what are known as vaccinal antibodies. By themselves, you probably could not call vaccinal antibodies vaccines, but one could argue that the combination of vaccinal antibodies and the tumor immunogen serves as a vaccine generated endogenously. Indeed, such immune complex vaccines have a long history. CAR T cells are another complex case: these are patient T cells engineered to express a chimeric antigen receptor (CAR) that targets whatever you assign it to target (but usually tumor antigens). The CAR T cells themselves are a form of passive immunotherapy, albeit a long-lasting one. However, CAR T cells themselves (along with other types of cancer immunotherapy) have been shown to give rise to epitope spreading in some patients wherein the immune system begins to target tumor epitopes not targeted by the CAR T cells, giving rise to immunological memory and helping to enable durable remission against the tumors. In that capacity, I would argue that CAR T cells can be considered vaccines under the same rationale as vaccinal antibodies. Reasonable people however might disagree here noting that the aforementioned epitope spreading is a bystander effect and thus not consistent with the design of a vaccine.
Anyway, the definitions I use here are probably less rigid than those used by regulators, but in my defense, under most contexts, rigid definitions are silly (for regulation it probably makes sense though because it affects who reviews the investigational new drug or device, and it’s important that it be the people with the most expertise about that thing). For example, organic chemistry is the study of the chemistry of carbon, but it wouldn’t have progressed nearly as far if organic chemistry had decided to shun everything except substances that have carbon in them. Our ideas for what things count and don’t count as part of a field will evolve as the fields progress- broad definitions ought to be embraced.
Antibody: A secreted Y-shaped protein (like a two-tined fork) that binds to epitopes via its paratope, acting to stimulate the responses of other components of the immune system. the terms immunoglobulin or B cell receptor (BCR) describe the same protein, but also include the membrane-bound form found on the surface of B cells.
It’s actually even more complex than that: no one is exactly sure which cells are the long-lived plasma cells (LLPCs). In immunology, different cell types are defined by various markers on their surface (often these get a very official “CD” in front of their name followed by a number, wherein CD stands for “cluster of differentiation” because the cells bearing that marker can be differentiated from other cells through flow cytometry and related techniques). There are no surface markers that can definitively separate out which cells the long-lived plasma cells from the short-lived plasma cells. We do know long-lived plasma cells are more likely to have certain markers on their surface, but they can be found on short-lived plasma cells too, so none of these are definitive. We can tell apart LLPCs and SLPCs by looking at their mRNAs, but this requires killing the cells. In essence, LLPCs so far have been defined by the fact that they are long-lived and also plasma cells. As summarized in a recent excellent review, a lot of what we thought about LLPCs in the past has been overturned in recent years:
It was once thought that LLPCs arise late into an immune response, but more recent work suggests that in reality LLPCs can emerge at any point in the immune response.
It was thought that the LLPCs were populated by cells with especially high affinity antibodies, but more recent work fails to confirm this.
It was thought that LLPCs were largely sessile, but they have shown dynamic migration patterns.
Valency: A confusing term that describes either how many binding sites an antigen has or how many different antigens are in a vaccine, depending on the contexts. For example, a monovalent (mono = 1) vaccine has a single antigen, such as the spike protein of a single variant of SARS-CoV-2. Alternatively, antigens can be displayed in a multivalent manner, such as on a virus-like particle (VLP), where each particle has multiple copies of the spike protein:
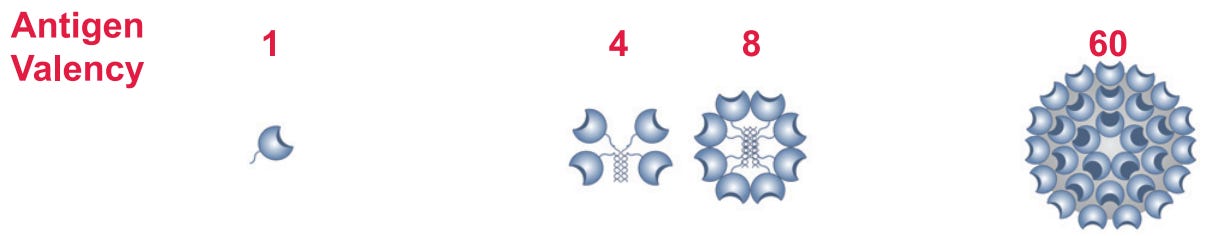
This means that a multivalent vaccine can also be a monovalent vaccine (I did not invent this jargon, please do not direct your very justified annoyance at me). Generally if people are talking about the number of immunogens in a vaccine though they will say “(number)-valent vaccine” whereas if they are talking about how the immunogens are structured they will use terms like “multivalent display.”
Immunogen: Anything that elicits an immune response. Often used synonymously with the term antigen- anything that the immune system can recognize.
The term immunogenicity describes the ability of something to elicit an immune response.
Epitope: The specific piece of an antigen that is recognized by an antibody or a T cell receptor (TCR). The part of the antibody or TCR that binds the epitope is known as a paratope.
Adjuvant: A substance added to a vaccine to increase immunogenicity and/or alter the profile of the immune response. Adjuvants can help reduce the need for additional doses, reduce the amount of immunogen needed in a dose, and broaden the immune response to cover more variants. Common adjuvants in vaccines include aluminum salts, oil-in-water emulsions, certain nucleic acids (not the ones in the COVID-19 mRNA vaccines- their modifications minimize any adjuvant activity), and certain lipids.
Infection: Invasion of a pathogen into a niche it is not supposed to occupy. Infections can be asymptomatic, paucisymptomatic (minimal symptoms), mild, moderate, severe, critical, or fatal.
Affinity: How tightly binding occurs for a particular interaction. The term “avidity” is a related one that is often used in immunology, and can be thought of as the sum of all of the binding interactions for a particular context. For example, you can have many low-affinity interactions come together to have high-avidity binding.
Immunological memory: the ability of the immune system to mount enhanced responses to antigens previously encountered and retain effectors from a prior encounter with the antigen. Memory can last potentially for the lifetime of the organism, or can be shorter-lived and is the result of the unique adaptations of memory cells (active immunity).
While immunological memory is generally required for protection against infectious diseases (and some other types of disease), it is not necessarily the same thing as protection, as it is possible for immunological memory to fail to be protective, or, in exceptional cases, worsen disease. Thus it cannot be assumed that a vaccine or infection that elicits an immune response will be protective without additional evidence.
Neutralization: The ability of an antibody to bind to and inactivate the thing it binds to, for example a viral protein or a toxin. Not all antibodies that bind to an antigen are neutralizing (but may still contribute to protection through other mechanisms).
Correlate of Protection (CoP): Something that can be measured that predicts protection (against a specific outcome) after vaccination. Theses are essentially always antibodies because antibodies are easy to measure. Correlates can be mechanistic, meaning that the thing you are measuring itself does the protection (i.e. it is the effector) or non-mechanistic (meaning the thing you measure is not itself doing the protection but it is strongly associated with the thing that is). Many people have a tendency to use the term correlate of protection to mean absolute correlate, meaning a threshold of the correlate of protection that predicts protection (such as a specific antibody titer). For example, we know spike antibodies are correlates of protection against SARS-CoV-2 through multiple lines of evidence (we even know they are mechanistic correlates), but we do not have an antibody titer we can measure and make judgments about protection, so we lack an absolute correlate.
Correlates of protection must always be defined with regard to a particular outcome. Possible outcomes include infection, colonization, transmission, disease, severe disease, hospitalization, death, etc.
Effectors: The components of an immune response actually doing the responding. These include antibodies, memory cells, cells of the innate immune system, and more. Similar to the idea of a mechanistic correlate of protection (assuming that the effectors actually are protective).
Sterilizing immunity: In the way most people mean it (today), an immune response that completely prevents infection. The concept is an ideal, but, under this definition, there is no vaccine or infection in all of history that has ever generated durable sterilizing immunity. Given that this is the case and the success of multiple vaccination programmes in controlling the spread of pathogens, sterilizing immunity is not a requirement for controlling transmission of infectious diseases as a whole.
A study has been done in mice to observe the effect of “flipping” expression of CD4 and CD8 on the T cells so that helper T cells have CD8 and killer T cells have CD4. CD4 allows T cells to recognize professional antigen presenting cells that have MHC Class II proteins that allow them to display peptides originating from outside the cell, while MHC Class I proteins are recognized by CD8 and found on all cells except red blood cells. The results is immune systems that are incompetent: rather than steering the immune response and helping B cells, the CD4 T cells kill whatever they interact with (including immune effectors actively dealing with infection) and the CD8 T cells cannot respond to exogenous antigens despite their helper phenotype because the cells they interact with cannot perform cross presentation (displaying exogenous antigens on MHC Class I).
Adaptive immune system: The part of the immune system that has to “adapt” to deal with specific threats through the production of memory cells. It works together with the innate immune system. It is generally the part of the immune system that we think of as having memory, although there is a different type of memory associated with the innate immune system known as trained immunity.
It is worth noting that B cell depletion in general will not completely eliminate all of the B cells or their descendants from the body. The LLPCs will tend to persist. It is very, very difficult to get rid of LLPCs, which can be a big problem if the LLPCs are the drivers of a disease, as they can be in some autoimmune diseases and cancer.
One of the factors complicating the study of HIV and vaccines for it is the limitations of animal models. Sooty mangabeys are a type of nonhuman primate, which one would intuitively think would reasonably recapitulate human immune function (as we are also primates). In fact, infection with SIV (simian immunodeficiency virus, essentially the monkey equivalent of HIV) does not result in AIDS in them because they retain double negative T cells (those not expressing CD4 or CD8) which compensate. These T cells do also exist in humans, but they are rare and clearly unable to compensate for the loss of CD4 T cells. African green monkeys also appear to be able to handle CD4 T cell depletion by SIV.
It should also be mentioned that T cell depletion alone does not explain the fatal effects of AIDS; there is a persistent immune activation seen in HIV which also is key for AIDS to occur.
This is actually even more complex than I described here (I know, you’re shocked). It has been found that TFH cells progress through discrete intermediates based on their expression of the transcription factor FoxP1 and the cytokine IL-21. TFH-full cells express low levels of FoxP1 and high levels of costimulatory molecules. It is therefore proposed that vaccines seeking to optimize antibody responses should emphasize development of TFH-full cells:

It should be noted that this is specific to a deficiency in the CD8 protein; there are other conditions which result in a deficiency of CD8 T cells but they also significantly affect other subsets of the immune system, which means the redundant immune mechanisms for protection cannot compensate as effectively. For example, mutations in the protein ZAP 70 may cause a deficiency in CD8 T cells with normal numbers of CD4 T cells, but this protein is an essential component of T cell signaling, including CD4 T cell signaling, which means that the CD4 T cells that are present are not functional (which in turn results in impaired antibody responses). Consequently, this immunodeficiency is considerably more severe than an isolated CD8 T cell deficiency.
Naive: Mature cells which have not yet been activated through exposure to the antigen (epitope) they are specific to. Naive cells have the greatest degree of stemness, but are much more needy in terms of their activation requirements compared with memory cells, which makes responses by naive cells much slower.
Inflammation: A set of evolved adaptations to recruit the immune system in response to a problem, and in some cases to deal directly with the problem. Inflammation is essential to our survival and whether it is good or bad depends entirely on the context in which it occurs. It is actually very difficult to define inflammation, so for interested readers I recommend this review.
Pathogen: A microbe (virus, bacteria, fungus, parasite) that causes disease. Whether or not prions technically qualify as pathogens is debated (also I highly recommend you read the acknowledgments on that paper for a laugh).
The existence of anatomical barriers can greatly complicate protection at mucosal surfaces with systemic responses. For example, in 2022, the Moseman lab showed that mice have a blood-olfactory barrier that restricts the ability of circulating antibodies to reach the olfactory epithelium. This is a problem not just because it means a susceptibility to respiratory infections, but also because it means a susceptibility to neuroinvasive infections, as the olfactory epithelium is actually able to reach directly into the central nervous system, circumventing the blood-brain barrier.

Protection at this site is mediated by a population of mucosal plasma cells which are induced through intranasal infections or vaccination and require T cell help. However, there are vaccine adjuvant strategies shown in this study to generate these mucosal plasma cells despite the intramuscular administration such as dmLT. The caveat here is that these studies were done in mice, and there might not be exact equivalence in the anatomic barriers here. Additionally, caution should be exercised with regard to the differences in infections across species and models, such as with COVID-19. While the neurological dysfunction observed in COVID-19 is well-recognized and can be debilitating, definitive evidence of infection of the central nervous system by SARS-CoV-2 has never been achieved despite exhaustive attempts to do so. Despite this, many mouse models of SARS-CoV-2 infection feature invasion of the central nervous system, including the brain (often because they express very high levels of human ACE2 and in inappropriate tissues).
In the context of immunology, systemic refers to the inside part of the body- things like blood, muscles, bone, etc. It is contrasted with mucosal, which refers to the parts of the body that have mucous membranes and are exposed to the outside word.
Any time you trigger a memory/recall response from the immune system, it is known as a boost. Despite that, the term “booster dose” has a more specific meaning. Booster doses are intended principally to enhance pre-existing immunity to some level after it has declined with the passage of time. Vaccines for influenza and COVID-19 are not technically boosters, as they are intended to cover different strains from those we have previously encountered- it goes beyond just a recalling of prior immunity.
Innate immunity: the generalized defenses of the immune system that are not specific to a given antigen. These include the complement system, pattern recognition receptors, anatomical barriers, and more.
Missense mutations change one amino acid to another one; nonsense mutations change an amino acid to a stop sequence.
I recall great ambivalence regarding my position on the need for boosters at the time. Mainly, I did not doubt that the boosters would markedly enhance protection against Delta, something that was eventually demonstrated unambiguously in a randomized controlled trial. The issue was that we faced profound vaccine inequity and those with 0 doses were in serious danger from this variant in particular- it seemed unjustifiable for the US to start administering third doses to people when so many still had none, not only from a moral position but even from the vantage point of self-interest. We would likely reduce transmission more by giving the first doses to people who had none than by giving third doses to people who already had two. However, the contracts that were in place for vaccine procurement in the US apparently did not allow for this to occur, which meant, regrettably, that the options were to either use the doses or waste them. Over a period of a few weeks, I pored over the data available to me and prepared to write up my assessment on my blog about the value and necessity of a third dose of vaccine, ultimately coming down on the position that it was more than a little unconscionable in the face of the inequity, but a third dose did stand to greatly improve our progress against the pandemic. About a day into writing this post, the emergence of Omicron was reported and everything I had spent time working to understand became irrelevant as it was undeniably obvious that a third dose would be needed.
I don’t know what exactly it is about these terms but when some people hear them, something weird happens in their brain and they hear “you are worse off vaccinated than not” when that is not at all what imprinting or the other terms mean. Imprinting means that the memory cells outcompete the naive ones in a particular immune response. In the worst-case scenario, this amounts to negative interference: the antibody response is less than what an individual who has not had prior experience with the antigen produces. Importantly, comparisons made here should be parallel. If someone has seen the ancestral spike protein 4 times and then sees an Omicron spike protein once, noticing that the response to the Omicron spike protein is smaller than the response to the ancestral spike protein tells you nothing about imprinting.
Not only can we make good antibody responses against Omicron -and we do- but Moderna even showed that boosting with their XBB vaccine eventually resulted in higher neutralizing antibody titers against the BA.5 variant than the ancestral variant in humans, demonstrating unequivocally that imprinting is not an absolute barrier to effective responses:

Even when looking at influenza, commonly thought of as the major pathogen of interest in the matter of imprinting, only about half of studies observe an effect of imprinting.
Reactogenicity: The common, expected symptoms after vaccination that result from local and systemic inflammation in the course of the immune response. Examples include arm soreness, redness, swelling, fever, muscle aches, joint aches, etc. Reactogenicity and safety are not the same thing. A vaccine can be highly reactogenic in that it frequently causes local and systemic symptoms while also being safe in that those symptoms remain self-limited and resolve on their own without resulting in harms to the health of the vaccinee.