



Is there DNA Contamination of mRNA Vaccines?
A critical look at some extraordinary claims and what they mean.

The gist: I recently made a post describing what we know about SV40’s ability to cause cancer in humans (the short version being: SV40 does not cause cancer in humans). The recent reignition of this panic about SV40 comes from a preprint that alleges the presence of a short sequence of SV40 DNA being present in mRNA vaccines, which has of course been inflated into “the mRNA vaccines are massively contaminated with DNA” and nonsense about how this means the mRNA vaccines will change our DNA. The preprint in question is, without exaggeration, some of the lowest quality science I have seen in the entire pandemic (I don’t think there is a single component of it that does not have critical errors). In truth, regulators constantly check each batch of vaccine for the presence of manufacturing impurities (including DNA and things far more concerning than DNA, like endotoxin) and have very strict, conservative limits on the levels of impurities that can be present for any vaccine that goes to the public (and furthermore data obtained illegally from the EMA through a cyber attack confirm that the levels of any residual impurities in the vaccine are far below established limits). Any vaccine batches that do not meet these standards are not released to the public.
I: Steelmanning the Argument
A warning: this part is long and technical because it requires me to go through a lot of cellular and molecular biology, specifically as it pertains to actual human cells and distinguish them from the cell strains and cell lines used in labs that have been adapted for specific applications. If you are interested specifically in the problems with the preprint, you can skip this part, but I do think it is valuable to show that every single part of this preprint could be accurate and it would still be meaningless.
Often I find it useful before assessing the merits of a paper’s claims to consider for a moment what it would mean if every single part of it were correct (this is absolutely not the case here). In large part, I’ve been a bit slow to address the claims here because they could be 100% accurate and it would not have any biological/medical relevance.
Let’s seriously consider for a moment what it would mean if there were high levels of DNA in the vaccines. Let’s even go as far as to say that this DNA is present within the lipid nanoparticles of the vaccine (something they didn’t show), meaning that the DNA would readily enter cells. What would that mean for our cells?
Some people have argued that even though we are exposed to DNA from all sorts of sources (food, our microbiome, literally any biologic medication- honestly everywhere) the presence of DNA within a lipid nanoparticle is unique to the vaccines and this therefore represents some sort of completely novel situation where the implications are totally unknown. This is complete nonsense.
I realize this might seem a bit silly to start with but it’s important to establish it early on: a mutation in one cell does not translate to that mutation in EVERY cell in the body, let alone germ cells (those that are used for reproduction). The cells that are descended from the mutant cell will have that mutation, unless they mutate that DNA again, but any cells not descended from that mutant cell will not have that mutation unless they also randomly acquire that mutation. Now then:
It is not simply foreign DNA that can (under a specific set of conditions) be a problem. For example, one of the major challenges genetically for all of our cells is managing gene dosage or copy number variation1. It is not just about having the right DNA, but the right quantities of the right DNA. There are a number of genetic conditions which are fundamentally problems of gene dosage. One of the most prominent examples is Down’s Syndrome, in which there is a third copy of chromosome 21 (trisomy 21)2. This copy of chromosome 21 is healthy. There is absolutely nothing wrong with it- but our cells are not designed to function with 3 copies of chromosome 21, and having this third copy causes a number of serious health issues. Down’s syndrome is in fact among the mildest case of these conditions (called aneuploidies) with the vast majority of them being lethal early in development. But what does Down’s syndrome have to do with lipid nanoparticles and extra DNA?

Every single moment you are alive, a bunch of your cells are dying (they are supposed to be- if they were allowed to live indefinitely they would eventually accumulate enough mutations to become cancerous). One of the pathways they can die is known as apoptosis, which involves the formation of structures called micronuclei. These micronuclei contain pieces of our genomic DNA, often quite large ones, and they are packaged into apoptotic bodies which are, essentially, a type of lipid nanoparticle containing genomic DNA. Apoptotic bodies can readily be taken up by our own cells, introducing DNA into our own cells and now reproducing the problem of gene dosage.

Except… they don’t create a problem with gene dosage. Why? For one thing, to create a problem of gene dosage, the DNA needs to transcribed. The enzymes responsible for this are found exclusively in the nucleus, and one does not simply go into the nucleus:

The cytosol of a cell is filled with DNases- enzymes that break down DNA. If you were to put a document into a shredder, it would be pretty hard to reassemble it into a coherent message. This is no different- DNA by itself can’t do much of anything3. It’s the contents of a DNA code that need to be read, interpreted, and acted upon. No message, no function. Hopefully it is starting to make sense why gene therapy approaches based on providing healthy DNA to a host are so challenging- but this is only a tiny fraction of the whole problem.
In reality, the microbes all around have ways to protect their DNA from degradation, so this would not be enough on its own. Fortunately, there is an entire additional arm to the detection of foreign DNA: the cGAS/STING pathway. DNA is essentially never supposed to be in the cytosol, and its presence induces the activation of a protein called cGAS (cyclic GMP AMP synthase) which does multiple things. Firstly, the cytosolic DNA is condensed, physically segregating it from accessing the nucleus. Then cGAS does as its name suggests and uses this DNA to create a metabolite called cGAMP. cGAMP in turn activates a protein called STING, which induces an antiviral state, which, eventually, results in the cell dying or being murdered by the immune system.

Amazingly, cGAS/STING is not the only way to kill a cell that has extracellular DNA. There are also a group of immune sensors called ALRs (AIM2-like receptors) which form complex structures called inflammasomes. Inflammasomes also (eventually) result in the death of the cell by either apoptosis or pyroptosis. Importantly, there are ALRs in the nucleus in addition to the cytosol, so if somehow DNA were to slip past all of these, it still would not escape the innate immune sensing pathways.
Of course, there are caveats here too. For example, when cells divide, they have to dissolve the nuclear envelope, in which case nuclear DNA is now in direct contact with the cytosol. If things during this part of the cell cycle acted as normal, then our cells would immediately shred any genomic DNA we had and it would be pretty hard to carry out life as we know it. Fortunately, we evolved a solution to that too and it’s actually very simple: heterochromatin. When cells divide, the DNA condenses extremely tightly to a form that is completely inaccessible to basically… anything (called heterochromatin)- including the enzymes that destroy or sense cytosolic DNA- or any extracellular DNA. Eventually, the nuclear envelope will reform and the DNA will relax into euchromatin in accordance with the cell’s transcriptional regulation wherein it will be able to carry out all the necessary functions for the cell’s survival.
Now, I already hear the naysayers- but what about this paper (or any other similar papers) showing spontaneous genomic integration of plasmid DNA in HEK293T cells?! Clearly this is a risk, and you’re just hiding it from us, Edward! Eh… no. HEK293T cells are a cell line that has been transformed using the T antigen of SV40 and is used for many applications, including the mass production of viruses and proteins for use in gene therapy and vaccines. This means that HEK293T cells have massive defects in fundamental aspects of the response against viruses- or else they would start producing interferon and shut off production, which would largely defeat the purpose of their use. As it happens, HEK293T cells are missing or have markedly reduced levels of: TREX1 (an enzyme that annihilates cytosolic DNA so as to avoid activating cGAS/STING pathway), cGAS, and STING. In other words, these cells are basically as incompetent as it is possible to for a mammalian cell to be to respond to extracellular DNA and not a reasonable model for what actually occurs in humans. But, since we are engaging in thought experiments here, @Berrytartlet did the math for us and according to this paper, assuming the regulatory limit of 10 ng/dose (which is far higher than anything found in the preprint by far), then the DNA in mRNA vaccines may cause 1 integration event in 1 cell. This is BEFORE you consider the fact that your cells are extremely hostile to any extracellular DNA and that HEK293T cells are rapidly dividing.
Yeah? Well what about the SV40 promoter, huh? I talked a great deal all about SV40 here, but I will reiterate: the totality of evidence we have indicates that SV40 does not pose a health risk to humans. Furthermore, the SV40 sequence in question here is not the virus, not even any T antigen, but a promoter sequence which (in bacteria) will (redundantly- more on that later) induce expression of a protein that allows the bacteria to survive in the presence of kanamycin. The DNA in question in the vaccine as reported by McKernan and colleagues furthermore is fragmented (because there is a step in the production process where it is digested) meaning it doesn’t encode any meaningful information. Even before it gets to the nucleus it still has to spend at least some time in the cytosol where it will risk destruction or induction of an antiviral state that kills the cell it has entered. If it somehow gets into the nucleus it still has the risk of the nuclear DNA sensors detecting it. The number of assumptions you would have to make for this finding to have any kind of biological meaning is astronomical.
Alright, but if DNA is so safe, why then are there regulatory limitations on how much DNA can be in any vaccine? As it happens, no one can quite agree on what the actual risks here are. In fact as Yang writes (emphasis mine):
It is not clear what health risk the DNA can pose in the product recipients, but often manufacturing can be designed to minimize the risk by reducing the levels of DNA.
The residual DNA in biologics like vaccines has never clearly been shown to be dangerous, but there was broad agreement that we should probably limit exposure to it. I can offer a few rationales here, but I would note that they are mainly theoretical. While DNA itself can’t do much of anything, if improperly modified (or not modified at all), it can cause inflammation and make for an unpleasant vaccination experience (there is in fact an adjuvant, CpG 1018, used in a hepatitis B vaccine that relies on the noxiousness of improperly modified DNA to the immune system to enhance immune responses to hepatitis B surface antigen; however, though it enhances the immune response to the HBV vaccine, it may reduce it to other vaccines depending on the type4). Another potential concern is the presence of retroviruses or other hidden genetic elements of concern in the cell substrate used to manufacture the vaccine being introduced- but this is not relevant to mRNA vaccines because they do not use cell strains or cell lines in their production. In other words, as Yang writes:
Interestingly, neither WHO nor FDA guideline requires manufacturers to conform to these limits; rather, as suggested in a WHO guideline (9), “a risk assessment should be done in order to define the DNA upper limit for a particular vaccine or biological product, based on the following parameters: nature of the cell substrate, inactivation process, the method used to assess DNA content, and the size distribution of DNA fragments.” Furthermore, the FDA also encourages manufacturers to discuss risk assessment and acceptable limits of residual DNA with the agency should an alternative approach or limits be used (10).
Put another way, even if what McKernan and colleagues suggest is true, it’s not clearly a violation of regulatory standards, and as I’ve hopefully made clear to this point, it poses no obvious safety risk either. The other potential risk alluded to is the presence of oncogenes within the cell substrate material that could be transported into the cell. In general, the cells that are used to make vaccines and other biologics tend to have many properties of cancer cells, and so there is a theoretical risk here that those oncogenes can get into the biologic. This one seems quite farfetched to me and the experiments suggesting it to be a risk are highly contrived (the oncogenes were flagged with long terminal repeats meaning they could be incorporated into the genome by retrotransposition), but it further lacks relevance here because there are no oncogenes involved at any step of production here and no cell lines or cell strains are either.
Lastly, and probably most importantly: the DNA in question is still vastly outnumbered by the mRNA in the mRNA vaccines (even with the nonsensical numbers the preprint proposes), meaning that any cell that takes up the lipid nanoparticles will all but certainly express the spike protein and die as a consequence of the immune response. If we could make cancers easily express potent antigens that our immune systems could target to destroy them, cancer would largely stop being a major public health problem. The idea that this represents some kind of genotoxic risk is ridiculous.
So basically, I’ve done my absolute best here to be as generous as I could be with the arguments claimed in this preprint as far as data, and I truly cannot find a good rationale that they would reflect a real health risk to vaccine recipients. However, I haven’t yet begun to enumerate the actual flaws in the work, some of which are truly astonishing.
The Risk of Insertional Mutagenesis through the lens of DNA Vaccines
Insertional mutagenesis is a general term describing a situation in which a piece of DNA is inserted inside another piece of DNA, thus changing the latter DNA’s sequence. Is there a theoretical risk of some kind of DNA inserting into the cell that takes it up? Sure- if you make many, many assumptions about all the mechanisms that have to go wrong for that to happen. It is however worth asking what the actual, practical risk here is that a DNA from outside a cell can just randomly insert itself into a genome. DNA vaccines have actually helped a great deal in answering these types of questions. In contrast to mRNA vaccines, DNA vaccines have to actually get inside the nucleus (because the machinery to transcribe the DNA exists only in the nucleus, and thus if they cannot reach the nucleus, they will not result in an antigen being made for the immune system to respond to). This has historically been the biggest barrier to the success of DNA vaccines. Because of the inefficiency of this process, the doses of DNA used for DNA vaccines tend to be very high. But, what actually happens to the DNA that gets taken up?
Turns out… it kinda just sits there (as the immune response gradually clears the cells that take it up). To quote from Ledwith et al:
Intramuscular studies in mice were carried out for three different DNA vaccine plasmids. We found that the plasmid persists in muscle tissue at least 6 months after injection, consistent with the observations of Wolff et al. [2]. Here, using our highly sensitive assay for integration, we showed that virtually all of the plasmid persisting in the muscle is extrachromosomal. Gel purification reduced the level of plasmid in the genomic down to ≤1–8 copies/ug DNA, and ≤1 copy/Ìg DNA in some cases.
…
Even if the residual plasmid in the gel-purified genomic DNA did represent integrated plasmid, it was previously calculated that 1 copy of integrated plasmid/ug of genomic DNA (representing 150,000 diploid cells) would be at least three orders of magnitude below the frequency of spontaneous gene-inactivating mutations [9].
In other words, integration events, even with techniques where the DNA is *designed* to get into the nuclei of cells, are extremely rare. In fact, the spontaneous rate of mutation is over 1000 times higher than the rate of these integration events.
Martin and colleagues obtained similar results (emphasis mine):
We have attempted to put this level of possible integration into perspective by making a calculation of the “worst case” scenario for an integration event in muscle cell nuclei and thereby a determination of the relative risk of injected plasmid DNA causing an untoward event. We can approach the question of risk by making a calculation, using the accepted spontaneous mutation rate of a single base in mammalian genomes as 1 x 10^-5 per gene (Cole and Skopek, 1994; Nichols et al., 1995). Since there is a significant loss of genomic DNA during our agarose gel separation method, the calculation is based on the mass of DNA assayed and not on total muscle DNA. Assuming the mouse genome contains ~3 x 10^9 nucleotide base pairs in 6 pg of DNA, 1 mg of genomic DNA represents the equivalent of 1.5 x 10^-5 genomes. This is the amount of DNA assayed in most of our PCRs. Using a further assumption that there are 7.5 x 10^-4 genes per genome, 30 integration events per microgram of genomic DNA would be equivalent to 2.7 x 10^-9 mutations per gene. Relative to a mutation rate of 1 x 10^-5 mutations per gene, the “worst case” mutation rate would be about 3000 times lower than the spontaneous mutation rate.
It is difficult to compare the risk inherent in all possible forms of mutations within genomic DNA, including the insertion of a plasmid DNA with eukaryotic expression control elements. However, the evidence indicates that plasmid DNA integration into muscle genomic DNA, should it occur, is such a rare event that it is difficult to quantitate, even using PCR. Without further evidence, plasmid integration must remain only a theoretical safety concern.
Hopefully this gives some sense of how difficult it is to do gene therapy at the level of DNA. The mere fact of getting an actual organism to take up the DNA in question is difficult, and you also have to avoid delivering it in such a way as to provoke the immune/inflammatory response that would interpret that message as a virus.
II: What’s actually wrong with the preprint?
A little bit of context here is helpful first. To make mRNA, you first need a DNA template. In this case, a circular piece of DNA called a plasmid is used. The plasmid contains the sequence for the spike protein (which will then be used for the RNA) and a selectable marker (in this case, kanamycin resistance). To make enough RNA, a large supply of plasmid is needed, and to make plasmid, it is easiest to insert it into bacteria and replicate those bacteria. To ensure that only bacteria that take up the plasmid are present, they are grown in the presence of kanamycin. Only bacteria that take up the plasmid should have the kanamycin resistance gene, so only they grow. Once they reach appropriate levels, the bacteria are killed and the plasmid is purified out and linearized (it goes from being circular DNA to having a clear start and end). From here, an in vitro transcription reaction is done with T7 polymerase and the nucleotides needed to make the RNA. The T7 polymerase is extremely specific to the spike RNA, i.e., it will not inadvertently wander off and start making RNA from other parts of the plasmid. That RNA is eventually purified and packaged into lipid nanoparticles.
It is not realistic that vaccines completely lack any trace impurities at all- the instrumentation to confirm the absence of even a single molecule of the impurities in question does not exist (and probably never will) and is not necessary. This is true of literally product we can consume. Nonetheless, vaccines (including mRNA vaccines) are subject to extensive purification steps and extremely conservative limits on what those trace impurities can be. In particular, the fact that the vaccine material of mRNA vaccines does not require the use of cells arguably makes them much “cleaner” than other types of vaccine.
In short, the preprint argues that vials of the mRNA vaccines contain unacceptably high numbers of DNA and unacceptably large fragments as well, but in truth even those claims are contrived. For example, here qPCR (the gold-standard method for quantifying DNA content) is used to calculate the quantities of DNA in the vaccines:

Red reflects spike DNA, blue is the plasmid’s origin of replication (the site where bacteria would start to make copies of the plasmid). Even with the use of a logarithmic scale here, it is blatantly apparent that the levels of DNA are much lower than regulatory limits. The preprint also shows that the DNA in question is highly fragmented, consistent with a digestion process during manufacturing (although you can reasonably argue that not all of it is < 200 bp long as regulators suggest- but more on that in a bit):
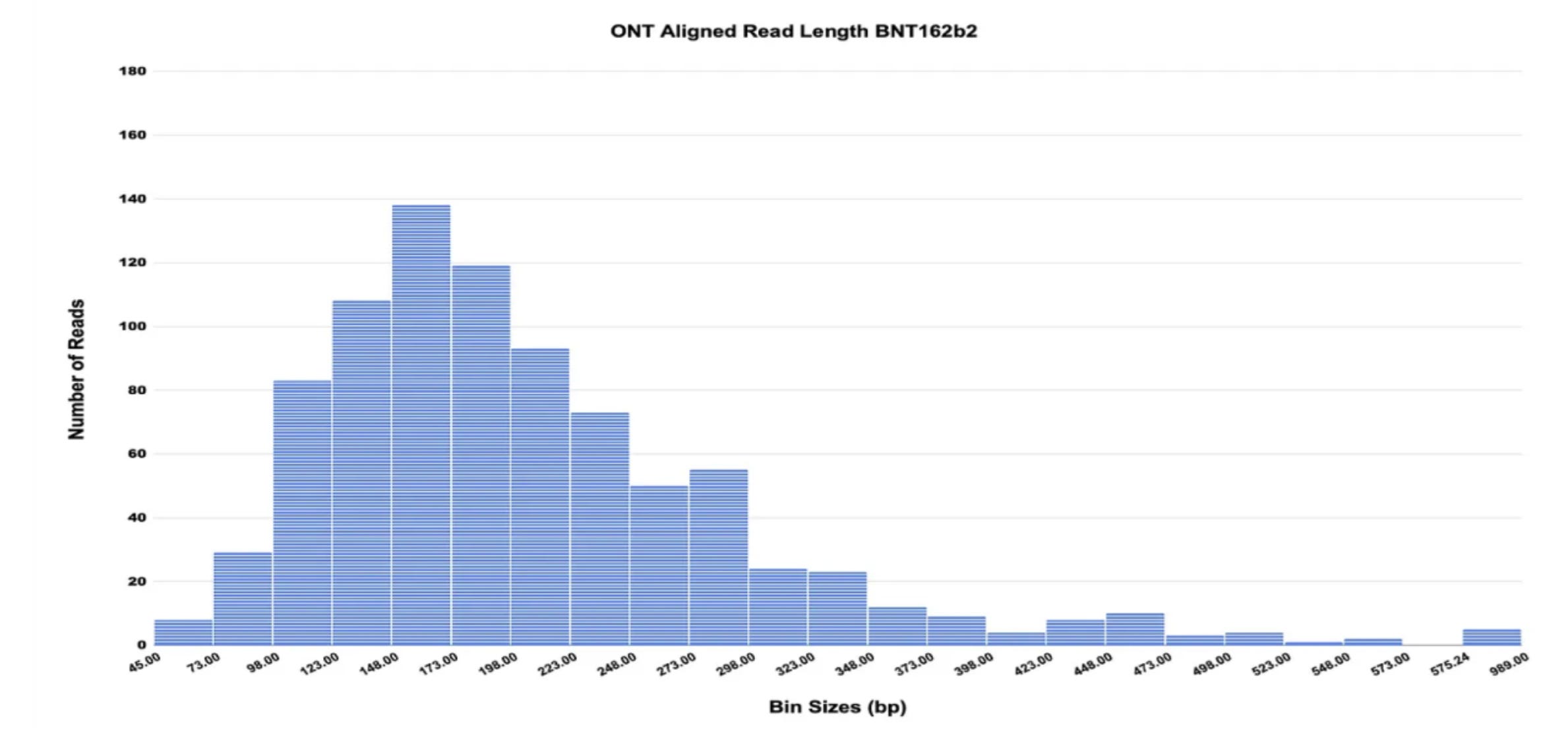
Perhaps most hilariously, the preprint attempts to allege a connection between the DNA content of the vaccines (by Qubit fluorometry- more on that shorly) and the risk of serious adverse events and it finds… an inverse relationship:

Yes, according to this, the more DNA in your vaccine, the less likely you are to experience a serious adverse event. So, how do you spin this non-data into a gripping tale of regulatory malfeasance and doom? You attempt to argue that these data actually aren’t the ones that matter and introduce non-data that you can claim proves the thing you decided to be true before doing any kind of investigation. Without further ado, let’s discuss the actual problems in this work.
Normally, I can extend an assumption of good faith to the authors of any given work of literature in science, but that is very difficult to do in this case. In truth, McKernan and his colleagues have an extensive history of publishing disinformation about vaccines and so this work should be viewed through that lens. One of the first and most glaring issues with the work is that there is no chain of custody apparent with the mRNA vaccines which were tested. In other words, there is no reasonable assurance that the products here have not been tampered with, and none of this work took place in a CLIA/GLP-certified lab. But, for the sake of argument, let’s let this slide (I know I’ve said that a lot in this post but there are a ton of things I’ve had to let go).
First let’s discuss the qPCR setup. In qPCR, you iteratively replicate a DNA sequence in a process guided by primers. These primers flank the sequence to define a start and end to the sequence of interest:

In the case of qPCR, you also monitor the progress of the reaction using a probe of some sort (there are a lot of variations on the type). As you make copies of your PCR product, the probe binds to it to produce a signal you can read (i.e., fluorescence). When it produces enough light to meet a certain threshold, the number of rounds of replication it took to achieve that is known as a cycle threshold or Ct (or Cq) value. The smaller the Ct value, the more starting material was present for your reaction. You can then use the Ct value to back calculate a mass for how much DNA was actually present. qPCR is a gold-standard technique for computing the amount of DNA in a sample.
The design of primers is actually not all that trivial in terms of the theory involved- although there are softwares that will readily do it for you in accordance with those principles. However, one of the biggest principles is that when you design a primer, it should be specific to a single part of the sequence if you want accurate quantification. The primer here is… not, as pointed out by Dr. David Mayhew:

The reverse primer for the SV40 sequence binds in 2 places and the forward primer may occasionally bind to a second site. This means that there are potentially 3 different PCR products that this reaction generates, meaning the DNA content cannot be accurately quantified. However… it is even worse than it sounds because not only do the primers bind in multiple regions of the SV40 promoter sequence… but, as our friend @BerryTarlet points out, the probe binds in multiple sites as well:

If the longest PCR product is made, then the probe will bind in both sites. Because the probe’s fluorescence dictates when the Ct value is met, having a probe that binds in multiple sites artificially lowers the Ct value and increases the quantified DNA. So, even though the qPCR allegedly showed that there is DNA from the plasmid that is far below regulatory limits, it is likely EVEN LESS than what the qPCR shows.
Of course, with the nonsensical qPCR data being inadmissible as evidence of the vaccine being dangerous, the team decided to use Qubit fluorometry. Qubit fluorometry is a technique which basically gives a rough estimate of the amount of a particular type of biomolecule (depending on the dye used) present in a sample. It does not, however, offer any sequence information. The argument offered is that some sequences are too short to be detected by the qPCR, and so this technique unmasks any DNA not captured by the qPCR. As I’ve already pointed out, the shorter a DNA sequence is the less information it contains and the less relevant it is biologically, so this is really grasping at straws. So, what’s the issue here?
RNA and DNA are very chemically similar. The dye used for qubit fluorometry is designed for the detection of DNA and it works well enough for situations where the DNA content is much higher than the RNA content (or even when they’re equal). However, the vials were not subject to digestion by RNases, so all the RNA is still present, and the RNA content should be far greater than the DNA content (if they were even close to similar, the manufacturing plants would never be able to make enough doses because the DNA template material would end up all being packaged into vials). The obvious interference of the mRNA with this quantification is apparent in the difference between Pfizer and Moderna vials. According to the qPCR, Moderna has much less DNA than Pfizer- but we know that Moderna has a much higher dose of RNA (100 mcg/dose vs 30 mcg/dose). Yet, Moderna’s Qubit data suggests far more DNA than Pfizer. Hmmmm… I’m going to go with “not likely.”
For the next part, I need to explain a bit about VAERS (the Vaccine Adverse Event Reporting System). Details on the US’s vaccine pharmacovigilance (the tools we use to monitor drug safety once they have been authorized to the market) mechanisms can be found here, but to give an abridged summary of the key points: the US has many safety monitoring systems for vaccines with varying levels of robustness. VAERS is one of the weakest in terms of robustness because it can be filled out by anyone, so the reports often contain incomplete, incorrect, or even fabricated information. Additionally, VAERS reports in isolation are not proof of a causal link between the vaccine and a given adverse event because to show that requires showing that the event is more common in vaccinated than unvaccinated people, and because VAERS only has vaccinated people, it cannot render those judgments. The point of VAERS is to act as an early warning system. Basically, if there is an unusual cluster of adverse events appearing in VAERS, it flags those events for interrogation in more robust systems like the VSD, which can make comparisons between vaccinated and unvaccinated people using their medical records and adjust for confounders. People should not take away that VAERS is useless- it’s not. VAERS has caught some really important safety issues in the past, and if something bad happens to you after vaccination, even if you don’t think it’s related to the vaccination you SHOULD report it to VAERS. The point of me explaining all of this is to explain that because VAERS exists in this government database and looks super official (and people like to ignore the disclaimer you need to confirm you’ve read to access VAERS), people have a tendency to misuse it to claim harms of vaccines that are not substantiated. For example, when Wakefield’s fraud about vaccines and autism first began, there was a massive spike in reports to VAERS about children developing autism. Millions of dollars in research funding and investigative journalism later, we know that there is no link between vaccines and autism, and that Wakefield’s work was fabricated so that he could sell his own measles vaccine (yes, really).5 There are other less extreme examples of outside events influencing VAERS. For example, when a vaccine first appears on the market, it tends to get more reports because people are watching it more closely, and as it becomes more established, these even out (this is called the Weber effect).
VAERS is foremost a US-based reporting system, but when I entered a selection of the lots discussed in the preprint (the ones with the most AEs associated with them), a large majority of the adverse events are reported as foreign. There is a requirement that if a serious and unexpected adverse event occurs from outside of the US, vaccine manufacturers report it to VAERS. However, when I restricted the query to just serious events (those that were fatal, caused hospitalization, congenital anomalies, or permanent disability), only about one-fourth of them met those conditions. When I further restricted to lots FN7934 and FM7380, however, there were only 21 serious adverse events reported for the two, whereas 77 adverse events were reported in total- this is starkly different from the reported 50% of cases which were serious adverse events. In any case, given the limitations of VAERS and the small numbers here, it’s hard to know what, if any, significance these numbers hold.
In addition to the actual data of the paper which are nonsensical, there are dishonesties in the claims it makes in the discussion of its findings. For example, it cites this paper stating that it claims that fragments as short as 7 base pairs have been known to spontaneously integrate in the context of DNA vaccines ignoring the full context of that remark:
In evaluating the potential harm of plasmid integration, it should be noted that the risk of introducing plasmids with strong regulatory regions into the host genome far exceeds that associated with random point mutations [43,50]. Moreover, the technology used to detect plasmid persistence does not examine the frequency with which short fragments of plasmid integrate. In this context, sections of DNA as short as 7 bp can affect rates of integration or recombination. Examples include the VDJ recombination signal sequence and related sequences, chi-like elements and minisatellites, ALU sequences, a recombinase signal present in hepatitis B and mammalian genomes, and topoisomerase II recognition sites [43].
In other words, the mere existence of a short stretch of DNA does not enable spontaneous integration unless it meets specific sequence requirements, and furthermore many of those requirements are cell-specific (e.g., VDJ recombination occurs only in the precursors to lymphocytes and thus they would be uniquely susceptible to integrating plasmids bearing recombination signal sequences).
In another instance, they suggest an association between the presence of DNA and the risk of ischemic stroke, citing this review. In reality, what the review says is that the conditions that put you at risk of stroke (chronic hypertension, hyperlipidemia, hyperglycemia) all result in mitochondrial dysfunction and release of mitochondrial DNA which can trigger inflammatory signaling via cGAS/STING and that cGAS/STING plays a role in the pathophysiology of stroke once it happens (which makes obvious sense given that a bunch of cells die and release their DNA). They do not address the effects of a sudden “bolus” dose of DNA as is implied to exist in this preprint.
Another example is related to the effect that DNA has on clotting. DNA is a long, negatively charged molecule which is the basic requirement for activating the intrinsic pathway of coagulation- but this gets to be a much lower risk as the length of the DNA shortens because there are fewer negative charges on it (are you starting to sense a theme?). There is reference made to a paper which describes what happens when endothelial cells are transfected with various formulations of DNA, which also demonstrate that the DNA induces a state that promotes clotting. The lipid nanoparticles containing the mRNA, however, are preferentially taken up by dendritic cells and other antigen-presenting cells. Furthermore, mRNA vaccines do not result in clotting, further weakening this position.
Lastly there were some more minor issues not directly within the paper that should be discussed but has been claimed from it. One is the claim that the presence of the SV40 promoter was concealed from regulators. This is untrue, which we know because regulators had the entire sequence of the plasmid. It may not have been labeled in the map of the plasmid, but that’s not essential or inherently deceitful, particularly given that it doesn’t encode anything. Another claim made outside this paper is that the SV40 promoter is essential for the production of the vaccine. The SV40 promoter is used to express KanR which allows for the bacteria to grow in the presence of kanamycin so long as they have (and replicate) the plasmid. However, here we are hoist by our own petard with this preprint because, although SV40 does show the ability to induce transcription in bacteria, upstream of the SV40 promoter is… an AmpR promoter (if McKernan et al’s results are to be believed).

III: What happens in the real world?
This has been a fun, if elaborate foray into a slice of science fiction masquerading as a preprint, but it makes you wonder what actually happens in the real world. Well, to help answer this we have the Rapporteur’s report from the EMA (European Medicines Agency) which was obtained through a cyber attack and made public discussing aspects of the Pfizer vaccine’s production. To the best of my knowledge, there is no analogous document publicly available for Moderna, but it is probably roughly similar. It is a very long document, but here are the important bits.
Initially the vaccine was made through a Process called Process 1, which had the problem of not being scalable to vaccinate billions of people, so some small tweaks were made to give Process 2. These really aren’t that different from one another. Basically, initially the DNA template for the mRNA vaccine was done on a PCR product. Unfortunately, this method is not going to give you nearly enough DNA template to make the RNA you need for the scale of vaccination to be done. So, a change was made to shift to a linearized DNA plasmid as I described earlier. Additionally, magnetic bead purification was not scalable, so instead ultrafiltration/diafiltration was used to get rid of basically anything that wasn’t the RNA. Furthermore, a Proteinase K step was added to break down any proteins that may have remained between steps.

Here’s an overview of the process:
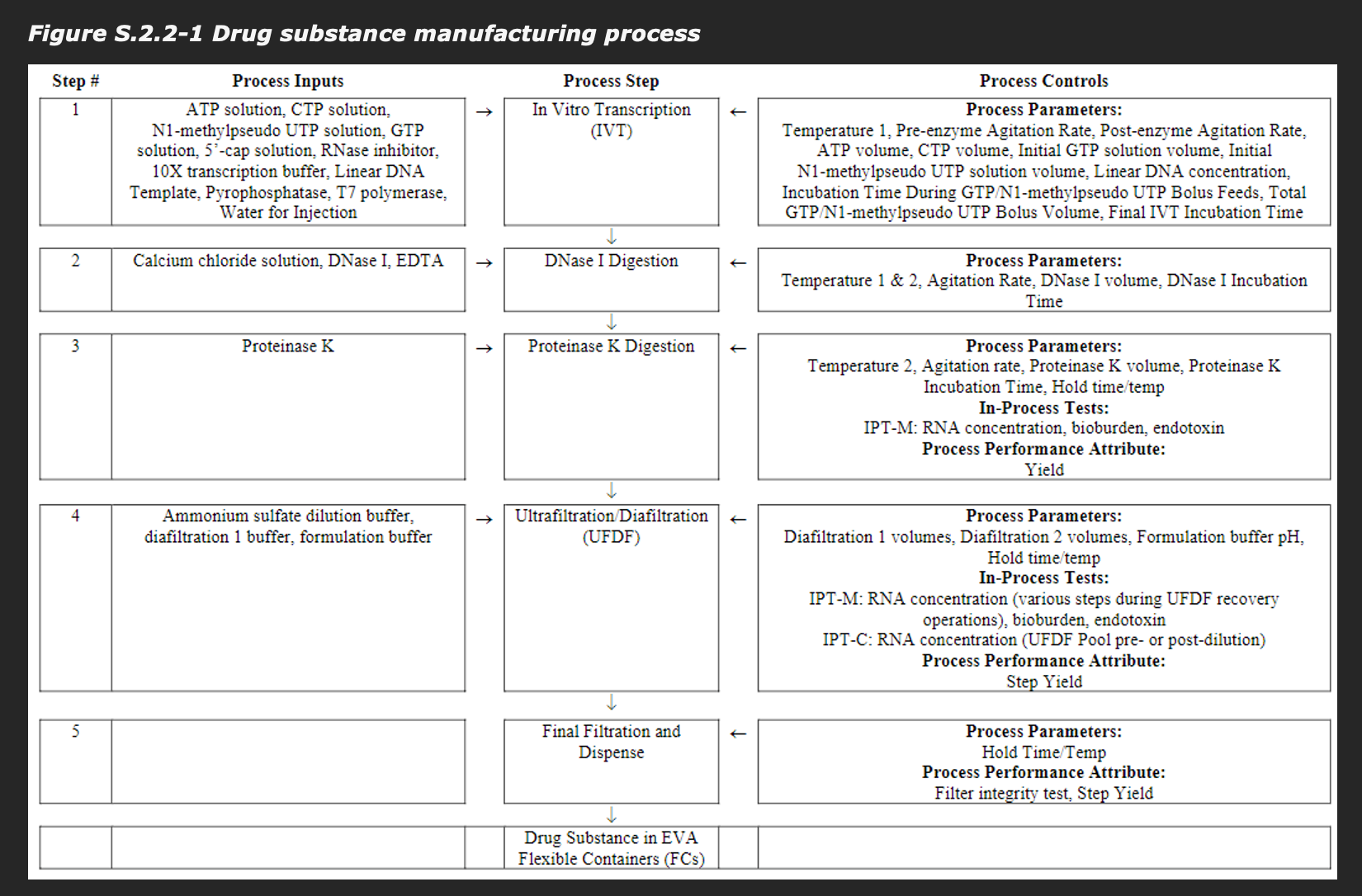
Note that there is a DNase I digestion right after the in vitro transcription reaction, reducing the amount of DNA and reducing the size of the DNA. As it happens, the EMA did perform testing of the residual DNA content of the batches and…

They are FAR below the acceptance criteria. There are other process-related impurities that are arguably more important, such as double-stranded RNA:

and especially critically, endotoxin:
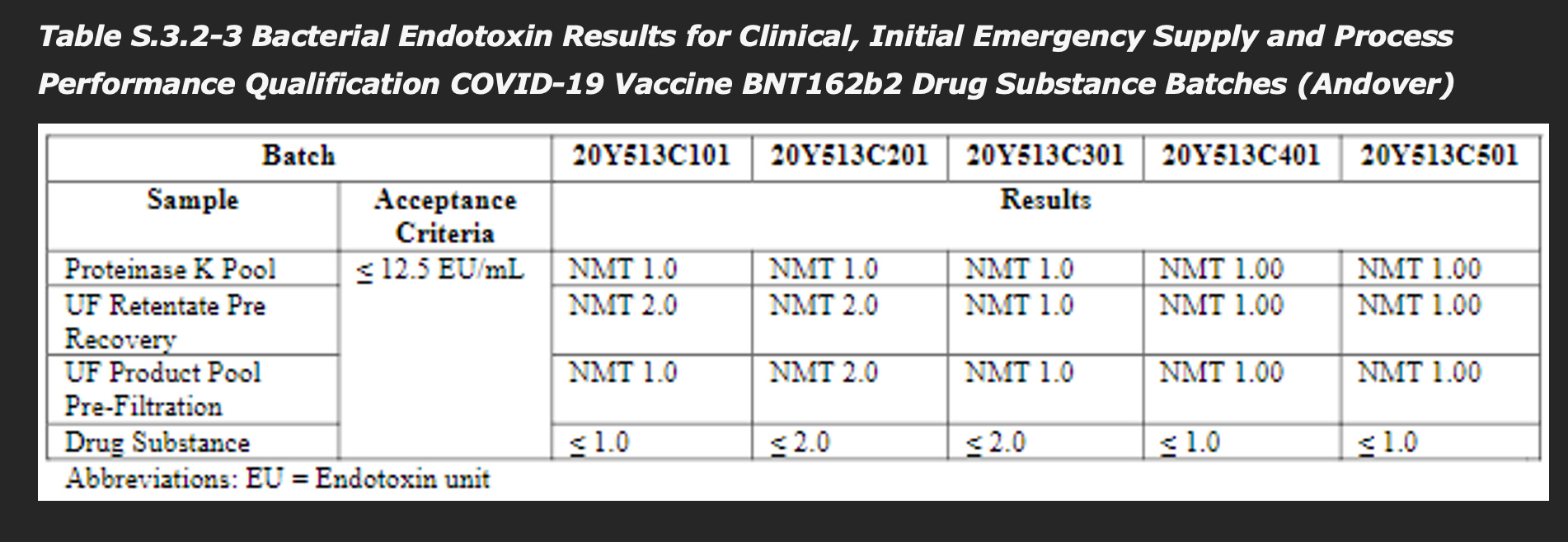
The report writes (emphasis mine):
Residual DNA template is a process-related impurity derived from the linearised DNA template added to the in-vitro transcription reaction. Residual DNA template is controlled by qPCR as defined in the DS specification, and the levels for all five batches are demonstrated to be well below the acceptance criteria.
Other regulators have offered comments:
The EMA spokesperson added that in the European Union, these results must be checked by an independent laboratory. “As a result, we are confident that the DNA levels in the vaccine are consistently below the approved/safe level,” the spokesperson said.
A spokesperson from the Therapeutic Goods Administration, which regulates medical products in Australia, told us that the agency has been monitoring batches of Moderna and Pfizer/BioNTech mRNA COVID-19 vaccines. “This includes independent testing performed by the TGA laboratories to confirm that residual DNA impurity levels are below the acceptable limit,” the spokesperson told us in an email. “To date all batches of COVID-19 vaccines supplied in Australia have met all quality specifications.”
Health Canada also chimed in:
“The SV40 promoter enhancer sequence was found to be a residual DNA fragment in Pfizer-BioNTech COVID-19 vaccine," the agency told AFP. "The fragment is inactive, has no functional role, and was measured to be consistently below the limit required by Health Canada and other international regulators."
Conclusions
Concerns about the DNA content of mRNA vaccines are unfounded and lack any reasonable basis.
Many people’s heads automatically go to cancer when they hear about mutations, but it’s a bit more complex than that. Cancer refers to a situation in which a cell begins to divide in an unregulated manner that does not consider the biological cues being provided to it, eventually forming tumors (usually- cancers of the blood cells are a bit different, although it depends on which one). However, getting cancer is not as simple as a single mutation. If you look at the DNA from a cancer cell and compare it to the DNA in a healthy cell, it is often described as though a bomb went off in the nucleus. Fundamentally, cancer is also a problem of gene dosage based on two types of genes: tumor suppressor genes and proto-oncogenes. Proto-oncogenes are genes that tell cells to progress through the cell cycle (i.e., they accelerate progress to cell division), while tumor suppressor genes delay progress through the cell cycle. Proto-oncogenes can have mutations that cause them to gain function so that they induce progress through the cell cycle more quickly (at which point they become known as oncogenes). However, cancer cannot occur until there is a mutation in BOTH copies of a tumor suppressor gene (remember- you get one copy of each of your genes from each of your biological parents, with some specific exceptions). Because you need both copies to be defunct, the odds of cancer occurring from any isolated mutation in the cell is exceptionally low, and so we again return to the issue of gene dosage: cancer (may) occur when the (effective) dosage of tumor suppressor genes falls below 1 functional copy.
Still, we should bear in mind that cancer is not just about what happens in the mutated cell but the body’s response to it (or lack thereof). The immune system remains a key player in the prevention of cancer, eliminating cancerous and precancerous cells every day. Problems being to arise when tumors get to a stage advanced enough that they can start to evade the immune system. This furthermore helps explain why cancer tends to be a disease of old age: in addition to the accumulation of DNA mutations throughout the lifetime in your cells, your immune function also declines dramatically as you get older.
In short, the development of cancer is complex and not as simple as “mutation *poof* cancer!”
A third copy of chromosome 21 (trisomy 21) is the most common way to develop Down’s syndrome, but it is not the only way. In a small proportion of cases there is a translocation where chromosome 21 is stuck onto another chromosome (usually chromosome 14) which results in extra replication of this chromosome before cell division and also reproduces the problem of gene dosage (this is called translocation Down’s syndrome). Another possibility is that rather than have the third copy of chromosome 21 be present from the moment of fertilization, the third copy may arise from an very error early on in development called nondisjunction, in which one daughter cell ends up with 3 copies of chromosome 21 and the other with 1. This results in all cells descended from the 3-copy cell having 3 copies of chromosome 21 and having the characteristics of Down’s syndrome, whereas those not descended from this cell division are healthy. This is known as mosaic Down’s syndrome.
The presence of DNA inside a cell that lacks appropriate modifications can be sensed by our cells and is generally interpreted as indicative of infection by a DNA virus. This induces an antiviral state within the cell and leads to a series of adaptations that typically end in the death of the cell- but more on that later. In this sense, I suppose you could argue that DNA itself “does something” but it’s more that DNA is sensed and it triggers a response rather than the DNA itself doing anything.
Specifically, because CpG DNA and cytosolic DNA in general induce type 1 interferon, when combined with live attenuated viruses or mRNA vaccines, they can suppress production of the antigen of interest within your own cells and reduce the antibody response. Vaccines which do not rely on these mechanisms however should be fine. Additionally, there are DNA vaccines which literally attempt to insert a plasmid that gets into the nucleus to make the antigen (this is incredibly inefficient and requires insanely high doses -relative to other vaccine types- of DNA).
The story of Wakefield’s fraud is incredibly byzantine and I cannot, given the constraints of length I imposed with this post, do it justice. If you are interested in the details, no account is as definitive or thorough as that of Brian Deer in The Doctor Who Fooled the Word. Deer is the investigative journalist who actually uncovered many key aspects of the fraud.