



[Imported Post] MTHFR: Mostly Irrelevant
Originally Posted April 4 2021

I am working on consolidating all of my posts on one site (substack) but unfortunately I have to do this manually as substack does not want to let me import my own materials automatically. This post reflects information current as of April 4, 2021.
The Short Version: MTHFR shows up a lot on the internet in discussions about adverse events after vaccination, but this is completely baseless. There has been one study ever that suggested there might be a relationship: it was examining the smallpox vaccine, which is not routinely given to anyone except members of the military (in case of a bioterrorist event) as smallpox has been eradicated and it in no way showed a definitive link between MTHFR gene variants and adverse reactions to the vaccine- and in fact its own authors don’t believe such a link exists. Further research on the subject has not shown a relationship between MTHFR variants and the risk of an adverse event following immunization. This makes sense- nearly half of the population has a variant of MTHFR where the enzyme is a bit slow (which is readily correctable by folate supplementation) but adverse events to vaccination are exceptionally rare. It just doesn’t make sense. Additionally, sacral dimples, stork bites, sugar bugs, and tongue ties are not markers of MTHFR deficiency- there is no evidence to suggest that they are. Despite this, the study has been used to justify completely inappropriate medical exemptions to vaccination, to the extent that the authors of the original study issued a commentary stating that their study was being misused for this purpose and should not be. Beyond this, the American College of Medical Genetics has a statement explicitly stating not to test for MTHFR variants (for individuals who are showing a tendency to form clots inappropriately- one of the known possible issues that may occur with certain versions of the enzyme, though the increase in risk is very small) because this test does not yield useful information (the primary reasons one might think to order this test would be a history of recurrent pregnancy loss or an increased tendency to form blood clots- further studies have demonstrated that MTHFR variants do not explain these conditions and therefore knowing a patient’s MTHFR status is not useful). Even though direct-to-consumer genetic testing companies will happily check MTHFR status, the reasons for testing are few and far between (in general it’s probably unwise to undergo genetic testing for medical purposes without the guidance of a medical genetics specialist like a licensed genetic counselor). MTHFR also has nothing to do with “detoxing,” or the metabolism of heavy metals or aluminum.
I: MTHFR and Vaccines

MTHFR shows up in pseudoscience du jour- for instance it’s the subject of a recent post from the inimitable Dr. Jen Gunter discussing a nonexistent connection between MTHFR and nonexistent estrogen disorders (watch closely- this will become a theme of the discussion), and also more recently here in relation to pregnancy loss infertility- but I really do think it got its start in vaccine pseudoscience. Les Witherspoon has a fantastic discussion of the gene and its relationship to vaccination here and I think it is more than adequate for people wanting to know more but I thought it would be fun to do a really deep dive (people have often remarked about my ideas of what constitutes fun- I know). The history of the matter is discussed in detail here, but here’s the relevant bit (emphasis mine):
This interest in MTHFR can be traced right back to Reif’s 2008 paper, which linked a variant of the gene to “adverse events” after smallpox vaccines. It was a somewhat intriguing result then. A decade later, however, James Crowe, the senior author of the paper and the director of the Vanderbilt Vaccine Center, offers a blistering assessment of his own study: “It’s just not even a valid study by today’s methodology.” To use it for granting vaccine exemptions now, he says, is “illogical and inappropriate.”
…
In the early days of genetics research, scientists looking at a small number of genes in a small number of people found that certain MTHFR variants were linked to a range of maladies: blood clots, cancer, heart disease, pregnancy complications. This seemed to make sense, as MTHFR codes for an important enzyme in the body. But as scientists went from looking at hundreds to thousands to hundreds of thousands of people, they realized many of those variants were extremely common, found in up to 40 percent of the population in some cases.
More importantly, those associations with various diseases just didn’t hold up in bigger data sets and with better statistical tools. They had been flukes all along.
To be frank, that’s basically all there is to the story of MTHFR in relation to vaccines. It was proposed as part of an exploratory study and further investigation showed it to be irrelevant, which happens sometimes- in exploratory studies most of the “hits” you generate will be false positives. The authors even went as far as to post this to tell people explicitly not to use their data in this manner.

There’s also a pervasive claim that MTHFR has something to do with “detoxing” but this is also nonsense. It is probably most instructive to discuss the real science of MTHFR. Unsurprisingly, MTHFR variants also do not have obvious physical signs contrary to popular belief- one of the most common claims on the interwebs is that children with the variants may have tongue ties (ankyloglossia), stork bite (nevus simplex), sacral dimples, or sugar bugs (a prominent vein over the nose)- there is no evidence to support that any of this is true.
First though a brief primer on terminology here: because this is a subject that involves genetics, people can be a bit sloppy in the way they discuss it (or sometimes deliberately misuse terminology) so we need to clarify some things. The American College of Medical Genetics and Genomics and the Association for Molecular Pathology state:
A mutation is defined as a permanent change in the nucleotide sequence, while a polymorphism is defined as a variant with a frequency above 1%. However, the terms “mutation” and “polymorphism”, which have been used widely, often lead to confusion due to incorrect assumptions of pathogenic and benign effects respectively. Thus, it is recommended that both terms be replaced by the term “variant” with the following modifiers: (1) pathogenic, (2) likely pathogenic, (3) uncertain significance, (4) likely benign, or (5) benign.
For simplicity, I will generally use the term “variant” throughout this post without qualifying it (doing so is complicated because the high abundance of variants in the population suggests some kind of selective advantage but what it might be is currently unknown; in that way the variants could be considered to have unknown significance but there are well defined circumstances in which they do pose risks e.g. neural tube closure). Also as a note on conventions: MTHFR = the gene, MTHFR = the enzyme.
The section on the biochemistry of MTHFR is technical and not necessary for understanding the role of the enzyme in health and disease for those who are uninterested, but is included solely for comprehensiveness and because my readership seems to be very diverse in interest level and background knowledge. Those seeking a thorough understanding should not omit the sections on the genetics and genomics of MTHFR in health and disease, however.
II: The Biochemistry of MTHFR (Warning: Technical)

MTHFR is a gene encoding the enzyme N5,N10-methylene-tetrahydrofolate reductase (MTHFR), an enzyme in folate metabolism (more specifically, in the methionine cycle) that catalyzes the formation of N5-methyl-tetrahydrofolate (N5-methylTHF) from N5,N10-methylene-tetrahydrofolate (N5,N10-methyleneTHF; this reaction requires an FAD cofactor), which is the rate-limiting step in the methyl cycle. Folate is an important player in one-carbon metabolism, a set of biochemical reactions in which single-carbon units are added or taken away from substrates, where it functions specifically in the transfer of one-carbon units of intermediate oxidation levels (carbon dioxide is transferred by biotin and vitamin K to perform carboxylations and most methylation reactions depend on S-adenosylmethionine) corresponding to those equivalent to methanol, formaldehyde, and formate. One-carbon metabolism is highly compartmentalized between the mitochondria and the cytosol and plays roles in biosynthesis (purines and thymidine), amino acid homeostasis (principally glycine, serine, and methionine), epigenetic maintenance, and redox defense, and in the mitochondria is also responsible for producing NADPH and glycine. The term “folate” describes a group of molecules (there are about 150 of them known and experts debate whether or not there are others yet to be discovered) that are all known otherwise as vitamin B9 (which is water soluble). They are defined by a common core structure involving three parts:
a pteridine ring that can be reduced or oxidized (this rarely plays a role in the chemistry of folates, with the notable exception of the synthesis of dTMP, shown in black in figure 24.9)
a para-aminobenzoic acid (PABA) linker that together with the pteridine ring binds 1C units (shown in pink in figure 24.9), and
a variable chain length polyglutamate tail that serves to localize the molecule within the cell (shown in blue in figure 24-9). This linkage helps to ensure folate gets trapped within the cell with more linkers resulting in additional negative charges that make egress more challenging.
Single carbon units can be added to N-5, N-10, or both (labeled in green in Figure 24.9), which is the principal difference between the folates, as well as the oxidation state of the pteridine ring. Mammals can synthesize the pteridine ring component but require diet to obtain the PABA linker and the glutamates. Tetrahydrofolate is shown in Figure 24.9, and it can be obtained from either diet or as a metabolite from the gut microbiota. Folates are extremely diverse in terms of their chemistry because of the wide breadth of oxidation levels their single-carbon units can carry (see Table 26-1) and the role that these single-carbon transfers can have in biology generally. Broadly, folates can serve as acceptors of one-carbon units in catabolic processes and sources of the one-carbon units for anabolic processes. Principally, folates are required for the synthesis of purine bases, the production of thymidylate for DNA (which is an unusual reaction because thymidylate synthase alters the redox status of the pteridine ring within the enzyme rather than the one-carbon unit present on the cofactor as all other folate-dependent enzymes seem to do), and regulation of the active methyl cycle (also known as the methionine cycle).

The methylation potential of N5-mTHF is generally too low for most biosynthetic methylations and thus methyltransferase enzymes must instead rely on the methyl group carrier S-adenosylmethionine (SAM or AdoMet; I prefer the SAM convention) which is activated because of the presence of an soft (as per HSAB theory) sulfur atom in the molecule that enables the transfer of methyl groups. SAM is converted into S-adenosylhomocysteine (SAH) which is broken down into homocysteine and adenosine. Folates and SAM are closely intertwined with one another metabolically as in the activated methyl cycle (also called the methionine cycle), methyl groups from N5-mTHF (this is the product of the reaction catalyzed by MTHFR) are added to homocysteine to generate methionine and this reacts with ATP to generate SAM; SAM in turn is hydrolyzed into adenosine and homocysteine which can again accept a methyl group to form methionine. N5-mTHF is the most reduced form of folate, and it is broken down into THF by an enzyme called methionine synthase (MTR) which depends on vitamin B12 (one of only 2 reactions that vitamin B12 is known to catalyze in mammals); this enzyme has wide tissue expression. As this occurs, a methyl group is transferred from 5-mTHF to homocysteine to generate methionine. The SAM/SAH ratio is an important indicator of the cell’s methylation potential (and therefore a key determinant of the cell’s ability to methylate DNA for instance). A low ratio indicates methyl donor deficiency. There are about 35 reactions known in humans that require SAM to function as a methyl donor, including those that produce epinephrine, melatonin, creatine, and phosphatidylcholine.

MTHFR (the human ortholog) has several means of regulation; it is a multi-domain protein of 656 amino acids with a well conserved N-terminal catalytic domain comprising a β8α8 (TIM) barrel, a binding site for FAD and NADPH and the product N5-mTHF, and a C-terminal regulatory domain. The enzyme forms homodimers in a head-to-tail manner via its regulatory domain. The catalytic domain alone has previously been shown to be sufficient for the entire catalytic cycle, which occurs via a bi bi mechanism. Eukaryotic orthologs of the enzyme also contain a C-terminal domain which can bind both SAM (which produces slow inhibition) and SAH (which reverses the inhibition). There is additionally a serine-rich 35-residue N-terminal domain in humans which can be multiply phosphorylated with up to 10 sites identified. This phosphorylation appears to prevent the degradation of SAM to SAH while bound to the enzyme, and this makes the enzyme more sensitive to the inhibitory effects of SAM. This action is mediated distally about 300 residues away from the site of phosphorylation. The differences in the Ki of the SAM-bound phosphorylated and dephosphorylated protein are small (2–3 vs. 6–7 µM) but thought to have physiological relevance because of the concentration of SAM within the cell (1–3 µM). Structural studies suggest that SAM-mediated inhibition is the result of the loss of the FAD cofactor required for catalysis, and catalysis could be rescued in vitro via FAD supplementation. Note that at low SAM/SAH ratios (indicating low methylation capacity and therefore reduced ability to synthesize thymidylate, methylate DNA or other moieties), MTHFR becomes disinhibited to restore SAM levels, whereas at higher ratios, its activity is suppressed. The phosphorylation status of the enzyme allows for fine-tuning of its activity and links it to other key cellular pathways.

Examination of the metabolic pathway shows that MTHFR deficiency would result in elevated levels of homocysteine, which is thought to be the principal pathomechanism involved in the risks associated with MTHFR deficiency, though how elevated homocysteine causes disease is poorly understood (several mechanisms have been proposed but evidence is largely lacking for any of them). I would hypothesize that the toxic effects arise from competitive inhibition of methionine-dependent functions in the cell, though in some disease processes it seems that homocysteine functions more as a biomarker of a pathologic process rather than its mediator. Because of homocysteine’s link to several metabolic pathways, it is hard to disentangle the effects of reduced metabolic flux of the nutrient from the co-stalling of other important metabolic intermediates and therefore difficult to link it directly to a pathogenic effect. A detailed discussion of whether or not homocysteine is causative in disease processes or represents an epiphenomenon can be found here.
It is imperative to note however that this is not the only means by which homocysteine can be converted to methionine in humans:
There is a second reaction catalyzed by betaine-homocysteine methyltransferase-1 (BHMT-1; a zinc-dependent methyltransferase which appears to work by an ordered bi bi mechanism) which produces methionine and dimethylglycine from homocysteine and betaine (trimethylglycine)- this does not depend on a folate cofactor. The quaternary ammonium species choline and betaine contain 3 labile methyl groups, allowing them to readily convert homocysteine to methionine. Importantly, there is differential expression of the BHMT enzyme across tissues: in nervous system tissue, the reaction of methionine synthase, which is a B12-dependent process predicated upon the presence of 5-mTHF occurs and BHMT is believed to be absent. In the liver, however, this process readily occurs, as well as in the kidney, and lens cells of the eye. In rat liver studies, it is estimated that this reaction accounts for about 25% of the remethylation of homocysteine to methionine, but other studies suggest its function in the liver with respect to remethylation is about equal to that of methionine synthase. BHMT is thought mainly to function as a control point for regulating betaine levels.
A more recently discovered BHMT-2 was noted. Like methionine synthase and BHMT-1, it is a member of Pfam02574 and like BHMT-1, its expression is greatest in the liver and the kidney. It has 73% amino acid identity to BHMT-1. The reaction however is not the same one as catalyzed by BHMT-1 or MTR: rather, the methyltransferase uses methylmethionine as a methyl donor with the aid of a zinc cofactor to methylate homocysteine, producing 2 molecules of methionine per catalytic cycle. The relevance of this enzyme physiologically to homocysteine metabolism is not presently well understood.
While one-carbon metabolism is central to many important physiological processes, I feel that a detailed discussion of it is beyond the scope of this present post, so I will instead refer interested readers to reviews by Zheng and Cantley and Drucker and Rabinowitz (which I found to be invaluable references). One-carbon metabolism is a key source of formaldehyde within the body, and the cells can shunt this formaldehyde into nontoxic formate that can be used in the production of nucleotides.
III: MTHFR Genomics and Genetics in Health and Disease
The ACMG statement is extremely comprehensive and succinct on this subject, so I refer anyone interested in more details to read it. The MTHFR gene is present on chromosome 1 (specifically, it is mapped to 1p36.3). Variants which reduce the function of the enzyme (formally known as hypomorphic variants in the parlance of genetics) are common. One of the more common variants produces a “thermolabile” enzyme (meaning that on heating its activity is abolished- this fact is not physiologically relevant for humans but rather is based on studies where the enzyme is isolated and experimented upon), resulting from the single nucleotide change C677T. This produces a missense mutation: Ala222Val (where alanine is changed to a valine at the 222nd position in the protein) that decreases the activity of the enzyme (it is estimated that in homozygotes having two copies of the variant the enzyme has 30% of its normal activity), causing a rise in the levels of homocysteine (the reaction that converts homocysteine to methionine requires N5-methylTHF which is the product of the reaction catalyzed by MTHFR). Another common variant that also reduces the activity of the enzyme is A1286C which causes a missense mutation: Glu429Ala (where glutamic acid is changed to an alanine at position 429 in the protein); it is thought that the resultant enzyme is impaired to a lesser degree than the thermolabile variants (homozygotes are estimated to have 60% of the activity of wild-type). Population genetics studies suggest that it is rare for individuals to have both variants at once, and this is only generally observed as compound heterozygotes in trans (meaning one copy of chromosome 1 has one variant, and the other copy of chromosome 1 has the other, rather than a single copy of chromosome 1 having both with the other being wild type). The thermolabile variant is very common, even in a homozygous form (meaning it is present on both copies of chromosome 1) with >25 % of Hispanics and 10-15 % of Caucasians found to have it (though <1 % of African Americans do). A1298C is found in 7-12% of North American, European and Australian populations and is less common in Hispanics (4-5%), Chinese (1-4%), and Asians (1-4%). Reduced activity of the MTHFR enzyme (MTHFR deficiency) produces elevated levels of homocysteine in the blood and urine (homocysteinemia and homocystinuria) and the extent varies with the degree of deficiency; for these common variants, generally significant rises do not occur unless also coupled with a deficiency of folate from diet. Multiple epidemiological studies have suggested that this is linked to too many conditions for me to list, but the major ones would be thromboembolic diseases and neural tube defects, so I’ll discuss those in more detail. Fortunately, the mild to moderate hyperhomocysteinemia that results from MTHFR deficiency is readily correctable by supplementation with vitamin B6, vitamin B12, and especially folate. These common variants do reduce the activity of MTHFR, but they do not cause severe MTHFR deficiency. The amount of research on the different variants is literally dizzying (more than once source has described it as the best-studied gene in existence), as the OMIM entry shows, and there are severe MTHFR deficiencies that have been documented which are clinically important, but they are exceedingly rare.
That said, MTHFR variants have been associated with a number of disease processes, but on closer inspection, these findings generally appear to be specious (there are, of course, exceptions). I will focus here on just a few of them: neural tube defects, cardiovascular disease and clotting abnormalities (thrombophilias), and homocystinuria.
IIIa: Neural Tube Defects

Closure of the neural tube is supposed to occur at approximately 28 days post conception, and failure to achieve closure results in neural tube defects. Folic acid supplementation daily periconceptionally (typically defined in clinical trials as the period 4 weeks before until 8 weeks after conception) is critical in preventing neural tube defects. Folic acid is a synthetic form of folate that gets converted in the body to the active form of folate (tetrahydrofolate, THF) via the enzyme dihydrofolate reductase (the major reason folic acid isn’t present in the body in large quantities is because there is an energetic drive to reduce it to tetrahydrofolate). The principal reason it is used for supplementation rather than folates that are naturally occurring, is that most biologically active folates are unstable and break down rapidly when exposed to oxygen and in solution. While folates can absolutely be obtained from diet, especially from leafy green plants, liver, etc. (though these folates do have a hard time surviving cooking), supplementation generally requires folic acid because it is much slower to degrade outside the body and therefore more likely to last long enough to provide nutritional benefit. In fact, folic acid was initially isolated from cells presumably as an impurity because other folates degrade very rapidly in the presence of oxygen. In the US, beginning in 1998, folic acid fortification was mandated for the US food supply, and it is present in enriched cereal grain products to reduce the risk of neural tube defects. Folic acid supplementation levels should not be adjusted based on MTHFR results, per ACMG. For prevention of NTDs, 400 mcg of folic acid per day is recommended for all women of childbearing age with a tolerable upper intake of 1000 mcg from fortified foods or supplements; the US Preventive Services Task Force (USPSTF) recommends 0.4 to 0.8 mg folic acid daily for all individuals planning or capable of pregnancy; in individuals who are high risk for neural tube defects or those who have had a history of them, the recommended dose is instead 4000 mcg (4 mg) daily- MTHFR variants do not apply here. Food fortification alone, while shown to reduce the risk of NTDs, is estimated to contribute on average only 163 mcg of folic acid, which is below recommendations (this still appears to have significant protective effects however, but the levels are considered suboptimal). The mechanism by which folic acid prevents NTDs is not fully understood but as per Molloy et al, any of the following (or a combination of these) are possible:
1. Disruption of cellular nucleotide synthesis: Genetic variants may cause subtle deleterious changes in folate-dependent enzymes involved in these processes, thereby causing impaired DNA replication and repair.
2. Disruption of DNA or histone protein methylation, thereby altering methylation patterns that exert crucial epigenetic control over cellular protein synthesis.
3. Disruption of the generation or flux of one-carbon units between competing folate-dependent pathways: The availability of one-carbon units depends on both cytosolic and mitochondrial folate pathways that extract these units from simple precursors such as serine and glycine.
4. Disruption of homocysteine processing within the cell, thereby promoting toxic concentrations of homocysteine in the microenvironment of the embryonic neuroepithelium.
An alternative hypothesis relates to microRNAs. microRNAs are short RNAs which are known to be critical regulators of genes early in development, and they frequently lie adjacent to highly methylated parts of the genome called CpG islands. Under conditions of folate deficiency (note that this is NOT synonymous with having an MTHFR variant), the genome may be less methylated. The methylation of cytosines at the CpG islands is thought to be a key mechanism of microRNA regulation; it has been demonstrated experimentally that miR-222 expression is enhanced under such conditions and this suppresses DNMT-1 expression (a DNA methyltransferase) resulting in global lack of methylation that enhances expression of many genes, including β-catenin, resulting in enhanced proliferation and reduced differentiation of nervous system cells, preventing closure of the neural tube.
Yet another hypothesis notes that lysines (an amino acid) in histone proteins around which DNA is coiled (which function to control gene expression) can undergo homocysteinylation; when homocysteine levels are high, these histones can undergo excessive homocysteinylation and lead to reduced expression of Cecr2, Smarca4, and Dnmt3b which are all genes known to be important for neural tube closure.
Importantly, not all NTDs are folate-responsive, but supplementation is estimated to be able to prevent 50 to 70 per-cent, which is quite good (the point being that sometimes they happen and it doesn’t mean that anyone was irresponsible or did something wrong- much of health is not within our control which is why it’s critical to be proactive when it is).
The principal concern with folic acid supplementation generally has to do with concealment of vitamin B12 deficiency; vitamin B12 deficiency causes megaloblastic anemia, but this can be corrected by either B12 supplementation or folate supplementation. The use of folic acid as a supplement can therefore conceal the presence of vitamin B12 deficiency, which is problematic because in addition to megaloblastic anemia, B12 deficiency can cause neurological damage which may become irreversible if not promptly addressed. Some argue that using 5-methylTHF as a supplement instead of folic acid may be superior because it circumvents this risk (folic acid is converted to THF, which undergoes further conversion into metabolites used in DNA synthesis that corrects the megaloblastic anemia; 5-methylTHF is part of the methionine cycle but the conversion to THF is dependent on vitamin B12, and thus supplementation will not be able to conceal B12 deficiency). That said, this issue can be avoided altogether with multivitamins that contain both folic acid and vitamin B12, so this argument is not particularly convincing to me (plus it’s generally uncommon for patients with a B-vitamin deficiency to have just one B-vitamin deficiency, hence it’s most probable that a multivitamin would be used regardless). Specifically, for the prevention of NTDs, there is not adequate data on any other supplement than folic acid for their prevention; even though 5-mTHF supplements do raise folate levels in the body at least as effectively as does folic acid supplementation, evidence is lacking that 5-mTHF prevents neural tube defects (they might- we just don’t have the data to show it and for that reason this represents an unnecessary and inappropriate risk). Additionally, these supplements are often substantially more expensive. That said, 5-mTHF is the most common folate in food; if you find this evidence unconvincing, you are better off increasing your dietary folates and take folic acid with them rather than spend money on an expensive supplement of uncertain value and forgo what is known to work.
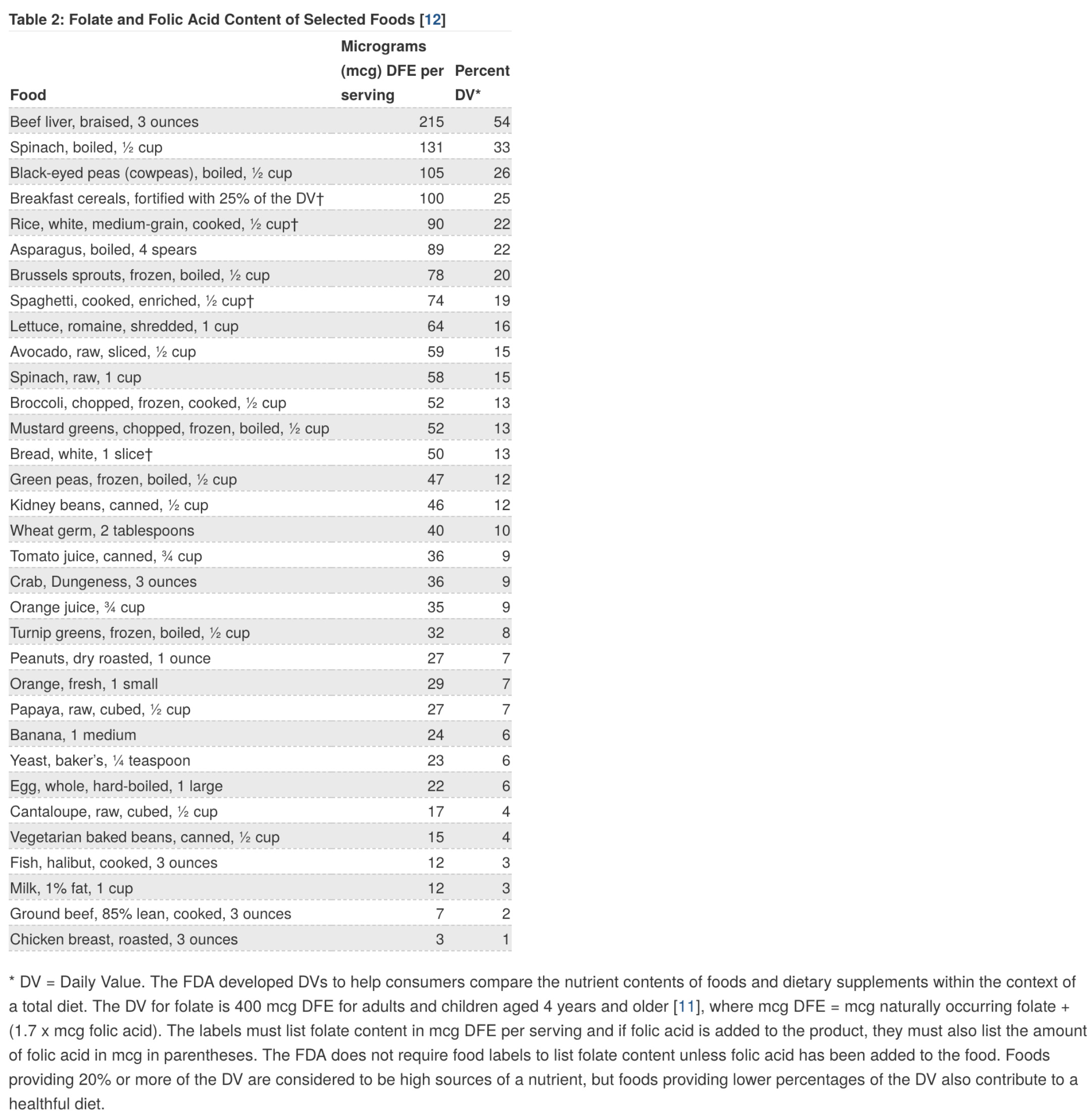
For those who have hypomorphic MTHFR variants, the prospect of 5-mTHF as a supplement seems attractive because it is directly providing the product of the reaction catalyzed by the enzyme and therefore appears to correct the deficiency more readily than does folic acid. However, data are clear that folic acid supplementation also reliably prevents neural tube defects even in those who have MTHFR variants (reviewed here). Though the activity of the enzyme is reduced in these variants, it is not absent, and it is known that cells only need catalytic (very small) quantities of folate for their needs (after all, guidelines recommend just 0.4 mg of folate equivalents daily, and some evidence suggests that benefits as far as prevention of neural tube defects occur at much lower levels). Of course, there’s also the fact that the variants do not exert nearly as dramatic an effect as many think:
When consuming the same amount of folic acid, people with the MTHFR 677 TT genotype have an average amount of folate in their blood that is only slightly lower (about 16% lower) than people with the MTHFR 677 CC genotype.2 However, researchers have shown that people with any of the MTHFR genotypes who consume 400 mcg of folic acid each day will increase (almost double) the amount of folate in their blood to an amount that is high enough to help prevent a neural tube defect in a developing baby.3 This means that your folic acid intake is more important than your MTHFR genotype for determining the amount of folate in your blood.
There is evidence to suggest that MTHFR activity may not be the key element to preventing NTDs (and thus supplementation of 5-mTHF may be no better than folic acid): Leung et al generated mice with total MTHFR knockouts (meaning no functional MTHFR is generated at all), and then bred them with each other so that both dams (the offspring) and their mothers where MTHFR-null (this way residual MTHFR activity in the mothers could not confound the results). Neural tube closure still occurred in these mice. This was explained by either the fact that the mice received adequate methionine (as inhibition of the methionine cycle did result in NTDs) in their diet or that the mice shifted their one-carbon source to generate methionine to be the BHMT (betaine-homocysteine methyltransferase) which also generates methionine from homocysteine and is a part of choline metabolism. In individuals who have MTHFR variants, this has been observed, possibly as a compensatory mechanism as well. The implication of this finding is furthermore that it is the formate- and folate-supported nucleotide production that is critical for neural tube closure, rather than MTHFR-mediated methyl group production. There is some caution warranted here though before extrapolating these findings to humans- we are after all not mice. Still, given how serious NTDs are (craniorachischisis and anencephaly are invariably fatal), I see no good reason to avoid a surefire option in favor of an unproven one, at least until data are gathered that prove it. Furthermore, Tsang et al.’s work suggests that the effects of MTHFR variants on increasing the risk of NTDs appear to be mediated by their effect on folate metabolism as a whole, rather than the remethylation of homocysteine into methionine, in which case 5-mTHF would not be predicted to have any greater value than folic acid among those with variants for the prevention of NTDs. There is additionally the paradox that the incidence of NTDs among non-Hispanic black women is lower than that of other groups despite them having lower levels of red blood cell folate further suggests that the relationship between folic acid, MTHFR, and neural tube closure is more complex than might be expected.
IIIb: Cardiovascular Disease and Thrombophilias

Homocysteine (and by extension MTHFR) have a complex relationship to cardiovascular disease- which is to say, one that is not at all obvious and therefore its measurement is of questionable value (like the ACMG has said). Moll and Varga have published an informative summary directed mainly at lay people. As described in Braunwald’s Heart Disease (emphasis mine):
Although homocysteine has also been linked to atherogenesis, prospective studies suggest, at most, a modest increase in risk associated with elevated homocysteine levels and have not consistently demonstrated a relationship independent of traditional risk factors or other biochemical markers. 10 Moreover, placebo-controlled trials have failed to demonstrate clinical benefit associated with folate and B vitamin replacement therapies as an intervention to mitigate the adverse effects of increased homocysteine levels. 11 Therefore, general screening for elevated homocysteine levels is not recommended.
…
A common polymorphism in the methylenetetrahydrofolate reductase gene (MTHFR) that encodes a thermolabile protein is also linked to elevated homocysteine levels and to increased vascular risk, at least among persons homozygous for the variant. Familial association studies have reported higher homocysteine levels in offspring of parents with premature CAD. The MTHFR polymorphism appears to have modest clinical importance, however, and heterozygous persons display little evidence of elevated homocysteine levels, even in those with low folate intake. In a meta-analysis of 40 observational studies, patients homozygous for the MTHFR 677 TT variant had a 16% increase in relative risk (odds ratio [OR], 1.16; 95% CI 1.05 to 1.28), and this observation was evident only in studies originating in Europe. 149 In populations in which folate fortification exists, such as in North America, little compelling evidence supports genetic evaluation of MTHFR to predict vascular risk.
…
Although elevated levels of fasting serum homocysteine (> 15 mmol/L) were once common, routine fortification of flour in North America with folic acid has lowered homocysteine levels in the general population. Elevated serum homocysteine may be associated with an increased risk for myocardial infarction, stroke, and peripheral artery disease, as well as VTE. Administration of folate along with vitamin B12 and vitamin B6 reduces levels of homocysteine. Nonetheless, randomized trials have shown that such therapy does not lower the risk for recurrent cardiovascular events in patients with coronary artery disease or stroke, nor does it lower the risk for recurrent VTE. Based on these negative trials and the declining incidence of hyperhomocysteinemia, enthusiasm for screening for hyperhomocysteinemia has declined.

In other words, at most, homocysteine levels (and MTHFR variants) have minimal importance in determining patient risk for cardiovascular disease, and given routine folate fortification in the North American countries, that value is further questioned. Furthermore, in randomized controlled trials the correction of the hyperhomocysteinemia does not appear to reduce risk of the cardiovascular events we are most concerned with, adding to the body of evidence that elevations in homocysteine are a (questionable) marker of risk rather than its cause.

The American College of Cardiology also seems less than enthused by homocysteine (emphasis mine):
Although there is an association of elevated homocysteine blood levels and CAD, a reduction in homocysteine levels with routine folate supplementation did not reduce the risk of CAD events in 2 trials (the NORVIT [Norwegian Vitamin Trial] and the HOPE [Heart Outcomes Prevention Evaluation] study) that included post–MI or high-risk stable patients (476–478) and produced poorer outcomes in another study (479). Additionally, in the NORVIT trial, there was a trend toward increased cardiovascular events (95% CI: 1.00 to 1.50; p = 0.05) in the cohort receiving the combination of folic acid, vitamin B6, and vitamin B12; the authors cautioned against using the treatment for secondary prevention (476). Similarly, experience in large clinical trials with antioxidant vitamins has failed to demonstrate benefit for primary or secondary prevention (474,475,480).
You can also see this from 3 separate guidelines that all direct against the use of B-complex vitamins including B12, folate, and B6 for homocysteine lowering as a method of preventing life-threatening heart attacks and other cardiovascular events or improving outcomes in patients with stable ischemic heart disease. It has been noted that in populations which have folic acid fortification of their food supply, the effects of MTHFR variants as far as homocysteine status and cardiovascular risk are inapparent, and this relationship is likely causal.
IIIc: Homocystinuria (Hyperhomocystinemia)

The presence of homocysteine in the urine (homocystinuria) indicates profound defects in the metabolism of the amino acid, as it is at that point building up to unacceptably high levels. This is serious and not the same as mild elevations in homocysteine that may occur with common MTHFR variants; there are MTHFR mutations which can cause huge rises in homocysteine levels that result in homocystinuria but they are extremely rare- about 40 cases have been documented ever. This condition is instead typically the result of a defect in a different enzyme: cystathionine beta synthase (CBS) (this form of the condition is sometimes called classic homocystinuria). This enzyme catalyzes a condensation reaction between the amino acids homocysteine and serine that produces cystathionine and water (or alternatively using cysteine instead of serine to make cystathionine and H2S), and is thought to act primarily within the nervous system but also in the liver and kidneys. Its activity is enhanced by S-adenosylmethionine (SAM), which is the major methylating agent in the body, whose production depends on the activity of MTHFR, and vitamin B6 is required for the reaction it catalyzes. From there, cystathionine is converted into cysteine, a critically important amino acid that can then be used to make glutathione and taurine; this is the only known pathway in the human body by which cysteine can be made. The incidence of homocystinuria is estimated to be 1 out of every 200,000 to 300,000 babies born in the United States, and is more common among those of Irish descent and children who are the product of inbreeding. There have also been cases of homocystinuria due to defects in methionine synthase, and there are 6 recognized forms that co-occur with methylmalonic acidemia resulting from defects in vitamin B12 metabolism.
Homocystinuria is very serious and for that reason is part of newborn screening. This is critical because children with homocystinuria appear healthy and normal at birth, but then go on to develop learning difficulties, nearsightedness, lens dislocation, epilepsy, osteoporosis, severe and premature atherosclerosis, and clotting abnormalities. The disorder may also cause folate deficiency (thought to be due to overconsumption of the folate pool from attempts to compensate for rising homocysteine levels). The condition closely resembles Marfan’s syndrome, a connective tissue disorder caused by mutations in the FBN1 gene encoding fibrillin-1. Death in childhood is possible, and if not screened for prenatally, the condition is usually detected only after permanent damage has resulted. It is fortunately amenable to treatment with dietary modification: supplementation with pyridoxine (vitamin B6) and betaine appears to help and a strict low-methionine diet must be adhered to. B12 supplementation may also be important depending on the underlying cause.
IV: Folate as a Nutrient and Supplement
The kinetics of folate absorption are complex. Folates must be present in their monoglutamated form for absorption, and fortified folic acid and supplements already are, but in foods, they are polyglutamated to facilitate intracellular trapping. Folic acid is notably about 85% bioavailable compared with 50% for natural folates. Thus folate hydrolase in the proximal small intestine cleaves off excess glutamates to form a monoglutamate. This enzyme depends on zinc and thus folate absorption can be hampered by zinc deficiency. Additionally, exocrine pancreatic insufficiency can increase the acidity of the proximal small intestine and reduce the activity of this enzyme. Once in the monoglutamate form, folates may be absorbed by any of several transporters on the enterocytes. The major one is the proton-coupled folate transporter (PCFT) present in the proximal jejunum and duodenum, and its function is optimized at a pH of 5.5-6.0. Within the large intestine, there is a reduced folate carrier which enables absorption of folates generated by the microbiota like 5-methylTHF. Once absorbed into the circulation, the folates move into the hepatic portal vein, exported by multidrug resistant-associated protein 3.
Once in the circulation, about 2/3 of the folates are bound to proteins including albumin, α2 macroglobulin, and a high-affinity folate-binding protein (with the remainder present in a free form). Most of the folate is in the form of 5-mTHF followed by 10-formylTHF, and in the plasma a typical concentration is 3 to 20 mg/L. There is actually more folate within the cerebrospinal fluid and red blood cells (taken up during erythropoiesis rather than by the RBCs) than in the plasma. Folate is taken up into the liver via carriers: RFCs (reduced folate carriers) take up folate into cells and PCFT is also present. organic anion transporting polypeptides (OATP) B1 and B3 (found on hepatocytes) also contribute. Folate receptor α (FRα) is important for folate transport into the central nervous system; deficiency of FRα is sufficient to cause neural tube defects. The liver is the principal site of conversion of folates into THF, which occurs with the aid of dihydrofolate reductase (DHFR) within the cytosol of the cell (this reaction requires an NADPH cofactor). Once in the liver, they undergo polyglutamation to trap them within the cell. They can also be released back into the digestive tract in bile and reabsorbed (enterohepatic cycling) almost completely. The body stores approximately 7 to 30 mg of folate at a time, most of which is in the liver, through the aid of folate-binding proteins and primarily in the form of THF and 5-mTHF.
Eventually, folate is excreted in the urine and feces. Folate-binding proteins in the kidneys transport the vitamin back into the circulation to retain folate if needed, but excess folate is lost and broken down into aminobenzoyl polyglutamate and pteridine. The folate that is secreted into the bile and not reabsorbed is lost in feces, though there is minimal loss by this route (though microbiota-produced folates may be lost more readily in feces).
As far as the balance of risks and benefits, supplementation of folic acid is extremely favorable, as the US Preventive Services Task Force’s most recent report shows. Most women do not receive the recommended daily intake of folate from diet alone, and furthermore about half of all pregnancies are unplanned, which together create an environment for susceptibility to neural tube defects. There really is no question that folic acid is very safe. Overall, the risks to supplementation are very small:
Masking of Vitamin B12 deficiency- easily avoidable through the use of a multivitamin that contains both B12 and folate
There are questions about the role of folic acid supplementation and the risk of colorectal cancer; these effects seem to arise only at very high levels of folate intake, and indeed at lower levels there may be a protective effect. The data are not very consistent on this point, suggesting that the risk is small if real at all.
Folates can interfere with antifolate medications (though sometimes taking antifolates may require folate supplementation e.g. methotrexate) and phenytoin.
Some studies have noted an increased risk of twinning with folic acid supplementation, but this effect is not consistently present and often is within the bounds of what might be expected from random chance alone.
Among pancreatic cancer patients, folic acid supplementation appears to worsen prognosis.
Though there is a theoretical concern about folic acid inhibiting dihydrofolate reductase at high doses, this has not been connected to adverse health outcomes thus far.
Because folates are water-soluble, they are essentially impossible to overdose on in the way that fat-soluble vitamins like vitamin D are. However, a tolerable upper limit for folic acid of 1000 mcg (1 mg) per day for women 19 years of age or older and 800 mcg (0.8 mg) per day for women 14-18 years of age has been set, but this is based primarily on the concern for masking B12 deficiency rather than the presence of toxic effects at higher doses, and there isn’t a defined upper limit for dietary folates (read: if you have an MTHFR variant and find the evidence thus far presented unconvincing, you can obtain N5-mTHF directly from diet as it is the most common dietary folate and take folic acid, but please do not forgo folic acid and be mindful of vitamin B12 levels; folic acid is about 1.7 times more bioavailable than natural folates). Indeed, 4 mg/day is recommended for those at high risk of having a child with neural tube defects or with a prior history of neural tube defects.
There are several ways to evaluate folate status. Red blood cell (RBC) folate levels are considered a diagnostic gold standard for evaluating folate status because they reflect a steady-state concentration of the vitamin (but for evaluation of folate status generally this is considered second-line); for the prevention of neural tube defect, folate levels of 1050–1340 nmol/L are considered optimal (note that they may be lowered by a vitamin B12 deficiency though). Serum or plasma folate can also be used for the evaluation of folate status, but these levels fluctuate more rapidly and reflect recent changes to folate status, and this is considered the first-line test. Thus for something like prevention of NTDs, RBC folate is the ideal metric, reflecting folate status for the last 2-3 months. Additionally, an alternative method to assess for folate deficiency is measurement of formiminoglutamate (FIGLU) levels in the urine, as FIGLU is converted to glutamate in a THF-dependent manner, but FIGLU levels rise with B12 deficiency as well. The same is true of homocysteine levels (but of course elevations can be due to deficiency of vitamins B6, folate, or B12). A final option is the deoxyuridine suppression test, in which the activity of thymidylate synthetase is measured in cultured lymphocytes or bone marrow cells (this reaction depends on the presence of folate and in more indirect ways on vitamin B12), and thus the enzyme will show reduced activity in the setting of folate deficiency that is rescued by the addition of folate (and similarly adding B12 for a B12 deficient individual would rescue the enzyme’s activity).
In short, anyone who is concerned about their MTHFR status almost certainly does not need to be. It is more worthwhile to instead check for homocysteine levels and act accordingly in virtually every circumstance and it has absolutely nothing to do with vaccination.
References
Molloy AM, Pangilinan F, Brody LC. Genetic risk factors for folate-responsive neural tube defects. Annu Rev Nutr. 2017;37:269–91.
Berg JM, Tymoczko JL, Gatto GJ. Biochemistry. 8th ed. New York, NY: W.H. Freeman; 2015.
Voet D, Voet JG. Biochemistry. 4th ed. Chichester, England: John Wiley & Sons; 2010.
Zheng Y, Cantley LC. Toward a better understanding of folate metabolism in health and disease. J Exp Med. 2019;216(2):253–66.
Les Witherspoon ND. MTHFR and vaccination: Much ado about nothing [Internet]. Ndsforvaccines.com. 2017 [cited 2021 Mar 27]. Available from: https://ndsforvaccines.com/mthfr-vaccination-much-ado-nothing/
Beale JM, Block J, editors. Wilson and gisvold’s textbook of organic medicinal and pharmaceutical chemistry. 12th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2010.
RCSB Protein Data Bank. 6FCX [Internet]. Rcsb.org. [cited 2021 Mar 27]. Available from: https://www.rcsb.org/structure/6FCX
Froese DS, Kopec J, Rembeza E, Bezerra GA, Oberholzer AE, Suormala T, et al. Structural basis for the regulation of human 5,10-methylenetetrahydrofolate reductase by phosphorylation and S-adenosylmethionine inhibition. Nat Commun [Internet]. 2018;9(1). Available from: http://dx.doi.org/10.1038/s41467-018-04735-2
Goyette P, Sumner JS, Milos R, Duncan AM, Rosenblatt DS, Matthews RG, et al. Human methylenetetrahydrofolate reductase: isolation of cDNA, mapping and mutation identification. Nat Genet. 1994;7(2):195–200.
Green R. Vitamin B12 deficiency from the perspective of a practicing hematologist. Blood. 2017;129(19):2603–11.
Stabler SP. Vitamin B12Deficiency. N Engl J Med. 2013;368(2):149–60.
5,10-METHYLENETETRAHYDROFOLATE REDUCTASE; MTHFR [Internet]. Omim.org. [cited 2021 Mar 28]. Available from: https://omim.org/entry/607093
Saldanha LG, Dwyer JT, Haggans CJ, Mills JL, Potischman N. Perspective: Time to resolve confusion on folate amounts, units, and forms in prenatal supplements. Adv Nutr. 2020;11(4):753–9.
Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25(1):27–42.
Mills JL. Preventing folate-related neural tube defects: Problem solved, or not?: Preventing Folate-Related Neural Tube Defects: Problem Solved, or Not? Birth Defects Res A Clin Mol Teratol. 2015;103(6):469–70.
Moll S, Varga EA. Homocysteine andMTHFRMutations. Circulation [Internet]. 2015;132(1). Available from: http://dx.doi.org/10.1161/circulationaha.114.013311
Final recommendation statement: Folic acid for the prevention of neural tube defects: Preventive medication [Internet]. Uspreventiveservicestaskforce.org. [cited 2021 Mar 29]. Available from: https://www.uspreventiveservicestaskforce.org/uspstf/document/RecommendationStatementFinal/folic-acid-for-the-prevention-of-neural-tube-defects-preventive-medication
Leung K-Y, Pai YJ, Chen Q, Santos C, Calvani E, Sudiwala S, et al. Partitioning of one-carbon units in folate and methionine metabolism is essential for neural tube closure. Cell Rep. 2017;21(7):1795–808.
Gunter J. The difference between folic acid, dietary folates, and so-called “natural food” folate [Internet]. The Vajenda. 2021 [cited 2021 Mar 30]. Available from: https://vajenda.substack.com/p/the-difference-between-folic-acid
Prepregnancy Counseling [Internet]. Acog.org. [cited 2021 Mar 30]. Available from: https://www.acog.org/clinical/clinical-guidance/committee-opinion/articles/2019/01/prepregnancy-counseling
Yan J, Wang W, Gregory JF 3rd, Malysheva O, Brenna JT, Stabler SP, et al. MTHFR C677T genotype influences the isotopic enrichment of one-carbon metabolites in folate-compromised men consuming d9-choline. Am J Clin Nutr. 2011;93(2):348–55.
Tsang BL, Devine OJ, Cordero AM, Marchetta CM, Mulinare J, Mersereau P, et al. Assessing the association between the methylenetetrahydrofolate reductase (MTHFR) 677C>T polymorphism and blood folate concentrations: a systematic review and meta-analysis of trials and observational studies. Am J Clin Nutr. 2015;101(6):1286–94.
Stover PJ, MacFarlane AJ, Field MS. Bringing clarity to the role of MTHFR variants in neural tube defect prevention. Am J Clin Nutr. 2015;101(6):1111–2.
Veeranna V, Zalawadiya SK, Niraj A, Pradhan J, Ference B, Burack RC, et al. Homocysteine and reclassification of cardiovascular disease risk. J Am Coll Cardiol. 2011;58(10):1025–33.
Zhou Y-H, Tang J-Y, Wu M-J, Lu J, Wei X, Qin Y-Y, et al. Effect of folic acid supplementation on cardiovascular outcomes: A systematic review and meta-analysis. PLoS One. 2011;6(9):e25142.
Fihn SD, Gardin JM, Abrams J, Berra K, Blankenship JC, Dallas AP, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease. J Am Coll Cardiol. 2012;60(24):e44–164.
Amsterdam EA, Wenger NK, Brindis RG, Casey DE Jr, Ganiats TG, Holmes DR Jr, et al. 2014 AHA/ACC guideline for the management of patients with non–ST-elevation acute coronary syndromes. J Am Coll Cardiol. 2014;64(24):e139–228.
Gerhard-Herman MD, Gornik HL, Barrett C, Barshes NR, Corriere MA, Drachman DE, et al. 2016 AHA/ACC guideline on the management of patients with lower extremity peripheral artery disease. J Am Coll Cardiol. 2017;69(11):e71–126.
Clarke R, Halsey J, Lewington S, Lonn E, Armitage J, Manson JE, et al. Effects of lowering homocysteine levels with B vitamins on cardiovascular disease, cancer, and cause-specific mortality: Meta-analysis of 8 randomized trials involving 37 485 individuals. Arch Intern Med. 2010;170(18):1622–31.
Zipes DP, Libby P, Bonow RO, Mann DL, Tomaselli GF. Braunwald’s heart disease: A textbook of cardiovascular medicine, 2-volume set. 11th ed. Philadelphia, PA: Elsevier - Health Sciences Division; 2018.
Ravanel S, Rébeillé F. Folate. In: Phytonutrients. Oxford, UK: Wiley-Blackwell; 2012. p. 173–202.
Rosenberg N, Murata M, Ikeda Y, Opare-Sem O, Zivelin A, Geffen E, et al. The frequent 5,10-methylenetetrahydrofolate reductase C677T polymorphism is associated with a common haplotype in whites, Japanese, and Africans. Am J Hum Genet. 2002;70(3):758–62.
CDC. Consuming enough folate helps prevent neural tube defects [Internet]. Cdc.gov. 2020 [cited 2021 Apr 3]. Available from: https://www.cdc.gov/ncbddd/folicacid/consuming-enough-folate-helps-prevent-neural-tube-defects.html
Mládková J, Hladílková J, Diamond CE, Tryon K, Yamada K, Garrow TA, et al. Specific potassium ion interactions facilitate homocysteine binding to betaine-homocysteine S-methyltransferase: Activation of BHMT by Potassium Ion. Proteins. 2014;82(10):2552–64.
Mládková J, Vaněk V, Buděšínský M, Elbert T, Demianová Z, Garrow TA, et al. Double-headed sulfur-linked amino acids as first inhibitors for betaine-homocysteine S-methyltransferase 2. J Med Chem. 2012;55(15):6822–31.
van Gool JD, Hirche H, Lax H, De Schaepdrijver L. Folic acid and primary prevention of neural tube defects: A review. Reprod Toxicol [Internet]. 2018; Available from: http://dx.doi.org/10.1016/j.reprotox.2018.05.004
Smith TC. Why you don’t actually need to test for MTHFR gene variants [Internet]. SELF. 2019 [cited 2021 Apr 4]. Available from: https://www.self.com/story/mthfr-genetic-testing-explained
Checkup Newsroom. A pediatrician goes in-depth into MTHFR [Internet]. Checkupnewsroom.com. Checkup Newsroom; 2020 [cited 2021 Apr 4]. Available from: https://www.checkupnewsroom.com/a-pediatricians-goes-in-depth-into-mthfr/
Lieberman M, Peet A. Marks’ basic medical biochemistry: A clinical approach. 5th ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2017.
Pajares MA, Pérez-Sala D. Betaine homocysteine S-methyltransferase: just a regulator of homocysteine metabolism? Cell Mol Life Sci. 2006;63(23):2792–803.
Levin BL, Varga E. MTHFR: Addressing genetic counseling dilemmas using evidence-based literature. J Genet Couns. 2016;25(5):901–11.
Litwack G. Human biochemistry and disease. 2nd ed. San Diego, CA: Academic Press; 2007.
Litwack G. Human Biochemistry. San Diego, CA: Academic Press; 2016.
Baynes JW, Dominiczak MH. Medical Biochemistry. 5th ed. London, England: Elsevier Health Sciences; 2018.
Walter JH, Jahnke N, Remmington T. Newborn screening for homocystinuria. Cochrane Database Syst Rev. 2015;(10):CD008840.
Sbodio JI, Snyder SH, Paul BD. Regulators of the transsulfuration pathway: Modulating the transsulfuration pathway in disease. Br J Pharmacol. 2019;176(4):583–93.
Newborn screening information for homocystinuria [Internet]. Babysfirsttest.org. [cited 2021 Apr 4]. Available from: https://www.babysfirsttest.org/newborn-screening/conditions/homocystinuria
Amoretti M, Amsler C, Bonomi G, Bouchta A, Bowe P, Carraro C, et al. Production and detection of cold antihydrogen atoms. Nature. 2002;419(6906):456–9.
Smith AD, Refsum H. Homocysteine, B vitamins, and cognitive impairment. Annu Rev Nutr. 2016;36(1):211–39.
Harrison’s principles of internal medicine. 20th ed. McGraw-Hill Education/Medical; 2018.
Zhang Q, Bai B, Mei X, Wan C, Cao H, Dan Li, et al. Elevated H3K79 homocysteinylation causes abnormal gene expression during neural development and subsequent neural tube defects. Nat Commun. 2018;9(1):3436.
Smith J, Gropper S, Carr T. Advanced nutrition and human metabolism. 8th ed. Belmont, CA: Wadsworth Publishing; 2021.
Field MS, Stover PJ. Safety of folic acid: Safety of folic acid. Ann N Y Acad Sci. 2018;1414(1):59–71.
Sobczyńska-Malefora A, Harrington DJ. Laboratory assessment of folate (vitamin B9) status. J Clin Pathol. 2018;71(11):949–56.
Hannibal L, Blom HJ. Homocysteine and disease: Causal associations or epiphenomenons? Mol Aspects Med. 2017;53:36–42.
Field MS, Kamynina E, Chon J, Stover PJ. Nuclear folate metabolism. Annu Rev Nutr. 2018;38(1):219–43.
Thanks Dr N for this review . I was always suspicious of the MTHFR fad in the early 00’s . I had many patients bring me their genetic testing results and wanted to take action about their MTHFR status. I sensed it was a bit of hype with our new found genetic analytic techniques. Now I understand more about it and have more to back me up as I try to reassure them. On another note- Did the article explain WHY Folic supplementation can mask B12 deficiency? I presume that fact has something to do with the linkage of THF and Methionine Cycles?