



[Imported post from November 11, 2022] Respiratory Syncytial Virus (RSV): A pathogen that will hopefully soon be vaccine-preventable
A one-stop guide to RSV infection, the history of old RSV vaccines, and what we know about immunity to RSV.

I am working on consolidating all of my posts on one site (substack) but unfortunately I have to do this manually as substack does not want to let me import my own materials automatically. This post reflects information current as of November 11, 2022. We have since achieved multiple safe and effective RSV vaccines for use in pregnancy and in older adults. What follows is the unedited form of the original blog post.
If you follow anyone in vaccine/medicine/immunology/infectious disease world, you may have heard some really exciting news that Pfizer has completed study of a candidate RSV (respiratory syncytial virus) vaccine showing high effectiveness against severe RSV disease in infants and the elderly. This follows GSK’s announcement of a highly effective RSV vaccine for older adults. You might reasonably wonder: Why is this such a big deal? Here’s the short answer: in vaccinology, there are some infectious diseases that are just incredibly difficult to make highly effective vaccines against, and historically RSV has been one of those pathogens, so this triumph marks a major advance in our ability to fight infectious diseases. Additionally, RSV causes some truly awful disease and is a major source of public health problems. The rest of this is… the longer answer. First though, some background (which will probably be enough for most readers who want to know about RSV; if you’re reading past the background, I’m assuming you’re a fellow nerd and will be treating you as such). I do think that for many people the last section will be of special interest, however, and so you may wish to skip to that part which I have attempted my best to put in plain language. The section on the History of the Lot 100 vaccine is also written in plain language and addresses why it has taken so long to get an effective RSV vaccine.
Background
RSV was discovered in 1955 in a colony of chimpanzees (known then as chimpanzee coryza agent) and causes seasonal outbreaks of disease every year (except for a period where it seemed to disappear related to precautions from the COVID-19 pandemic). It is not a new virus. Humans are the only known reservoir for RSV (don’t get too excited- it is likely never going to be eliminated or eradicated).
RSV is thought to be the leading cause of hospitalizations in infants with ~150,000 hospitalizations per year in infants and ~180,000 annual hospitalizations of the elderly from RSV. In 2019, 2.0% of deaths in children aged 0–60 months (UR 1.6–2.4) and 3.6% of deaths in children aged 28 days to 6 months (3.0–4.4) were attributable to RSV globally.
Another key risk group in addition to the extremes of age are those who undergo hematopoietic stem cell transplant or have hematologic cancers, have cystic fibrosis, or are otherwise significantly immunosuppressed.
RSV is mainly dangerous because of its ability to infect the lower respiratory tract to cause pneumonia and bronchiolitis.
Outside of key risk groups, RSV typically causes a mild upper respiratory infection akin to a cold.
RSV is the leading cause of viral middle ear infections (otitis media) in young children.
It has long been appreciated that children who have severe RSV infections early in life are at an increased risk of asthma, but it remains unclear whether the virus itself is a cause of asthma.
RSV regularly causes reinfections- nearly all children will be infected by the age of 2, and about half will have been infected twice. Lower respiratory infection (i.e., severe RSV) is rare for reinfections (due to acquired immunity)- it is typically mild (i.e. a cold) or inapparent.
RSV is generally thought to be spread overwhelmingly through direct contact or inhalation of virus-containing secretions at close ranges. However, modes of transmission lie along a continuum and a role for long range aerosols is possible, though it is not thought to be a major mode of spread for RSV. A detailed accounting of the appropriate infection control precautions against RSV can be found in this excellent thread.
RSV has a median incubation period of 4.4 days and generally falls within the range of 2 to 8 days.
Those infected with RSV are typically contagious for 3-8 days, though some shed virus for a while (weeks) after apparent recovery.
Asymptomatic (and presymptomatic) transmission of RSV is possible but it is not entirely clear how important it is to transmission. Asymptomatic individuals are less contagious than symptomatic individuals, but many RSV infections (especially in immunologically experience people) are asymptomatic, and thus it is possible that a large proportion of cases arise from asymptomatic transmission.
RSV season in the US typically begins in December, peaks in January/February, and ends by March/April.
The US CDC uses RSV-NET to conduct epidemiological surveillance of the virus.
RSV is genetically divided into two groups (A and B) based on antigenic properties (i.e., antibodies against A don’t, in general, cross-protect against B and vice versa- though in general all RSVs are considered to be part of the same serotype), with the most difference in the G and N proteins across the two groups. The F protein is relatively well conserved across the two groups, however. Compared to many other respiratory RNA viruses, RSV is not very genetically diverse, and instead seems to rely on immunomodulation to prevent the development of robust, protective immunity. Both groups of viruses typically cocirculate in a given RSV season, but one may predominate over the other. Some data suggest group A virus may cause more severe disease than group B virus, but these findings are not definitive.
RSV enters the cells via the action of its F, G, and SH proteins (but only F and G are required); neutralizing antibodies against F and G are protective. Antibodies against SH do not appear to neutralize but do accelerate viral clearance in animal models.
RSV’s F protein is metastable, meaning it readily interconverts between a pre-fusion and post-fusion form which makes generating neutralizing antibodies difficult because it conceals key sites on the pre-fusion F protein that can be targeted by neutralizing antibodies to prevent the virus from entering cells.
Monoclonal antibodies for RSV (Palivizumab) can be given preventively to some high-risk infants but these are very expensive and do not give perfect protection. Doses need to be repeated monthly for 6 months. AAP has now changed recommendations to allow for interseasonal doses given the change in the virus’s seasonality in 2022.
It is in principle possible to protect infants from severe RSV by maternal vaccination to transfer neutralizing RSV antibodies across the placenta.
More recent vaccine strategies have deduced ways to trap RSV’s F protein in the prefusion conformation so that neutralizing antibodies against it can readily be elicited. GSK and Pfizer both have completed trials of RSV vaccines which showed high effectiveness against severe RSV based on this strategy.
Historically, RSV has been an extremely difficult target because the first RSV vaccine attempt in 1967 was catastrophic: it not only resulted in a vaccine that was not protective but a vaccine that seemed to worsen disease to the extent that 2 toddlers died from severe RSV infection which has since been discovered to have been enhanced by the vaccine. This enhanced respiratory disease set back RSV vaccine efforts decades and is now a phenomenon that is closely watched for for any vaccine against a respiratory virus. It fortunately does seem to mainly be limited to inactivated vaccines of some respiratory viruses, but does NOT occur with any vaccine currently offered to the general public. Enhanced disease is also very obvious if it occurs in a clinical trial and with the size of current clinical trials, no vaccine showing enhanced disease would ever make it to market.
The current surge in severe RSV cases is most likely driven by the relative absence of the virus in prior seasons because of aggressive non-pharmaceutical interventions (NPIs) against COVID-19, causing a lack of boosting to RSV-specific immunity in the population. Normally, RSV immunity would be boosted by serial infections and this would limit its spread from season-to-season. If these infections do not occur, because there is presently no effective vaccine to rely on instead, there is a significant decline in RSV immunity in the population. This allows the virus to spread more efficiently and infect more people, potentially causing more severe cases as well (although direct evidence that RSV this season is also more severe, rather than there are simply far more cases and thus more of them are severe, is lacking). The solution to this is not to say that we should never rely on NPIs, but rather to redouble efforts for an effective RSV vaccine (which hopefully should be available for key populations by early 2023) and enhanced NPI use in the interim until the vaccine can be rolled out to significant numbers of people.
Most of the points discussed here will be elaborated upon in greater detail in the following sections (because people on Twitter had really excellent and detailed questions when I asked what they wanted to know), but suffice it to say, RSV is a very big deal in terms of public health, which would make an effective vaccine against it a massive triumph.
Clinical Manifestations of RSV and Risk Factors

RSV is similar to many other respiratory viruses in its signs and symptoms, and it is probably impossible to distinguish it from any of them by symptoms alone, though it is well known for certain disease manifestations in certain groups. Table 2 provides a list of some of the common manifestations seen in hospitalized and non-hospitalized children and proposes the global respiratory severity score (GRSS).
The first RSV infection is essentially always the worst one, as there is no baseline immunity to protect from severe disease manifestations, and usually occurs quite early in childhood. It can range from a mild cold to severe lower respiratory tract disease (occurring in 20-30% of first infections). Usually the upper respiratory tract infection period precedes the lower respiratory tract period by 2-4 days, and may be accompanied by a low-grade fever that often resolves by the point that lower respiratory tract disease occurs (but can also be intermittent and may be absent in very young children). Other common symptoms include rhinorrhea (runny nose) and congestion. A cough may occur with early upper respiratory tract illness as well as upon progression to the lower respiratory tract. Often, the disease does not progress into the lower respiratory tract phase, and most children will have lower respiratory tract disease only once (unless they develop reactive airway disease i.e. asthma). In infants, especially those younger than 12 weeks of age, a particular concern is the development of RSV bronchiolitis (inflammation of the bronchioles, the small airways). RSV is the most common cause of bronchiolitis in children, accounting for 50-80% of all cases of bronchiolitis. Is is thought that as the virus infects cells in the nasal turbinates, the cells are sloughed off and travel into the lower respiratory tract, carrying virus with them. In response to the infection, the lower airways enhance mucous production and sloughing of infected epithelial cells continues until eventually an obstruction may form causing atelectasis (essentially a collapsed lung) which prevents gas exchange for the alveoli below it (air trapping). Blood oxygen levels may decline as a result and the child’s breathing may get quicker (tachypnea). Children also develop a characteristic audible wheeze when infection progresses to the lower respiratory tract. Among children under age 5 hospitalized for community acquired pneumonia, RSV is the most common cause, and especially in children under age 2. Children younger than 5 months account for roughly two-third of all RSV hospitalizations during typical seasons, and prematurity in particular is a major risk (though it has been observed that the risk of severe manifestations of RSV are greater for those born after 29 weeks’ gestation than before) which is usually explained by chronic lung disease of prematurity (bronchopulmonary dysplasia). Infants younger than 4 weeks old may also develop apnea. For this reason, outbreaks in nurseries and NICUs are of particular concern. Other major risk factors include non-white race, low socioeconomic status, congenital heart disease, chronic respiratory disease, neuromuscular impairment, significant immunodeficiency, and Down syndrome. Interestingly, RSV does not seem to substantially exacerbate cystic fibrosis, though cystic fibrosis has historically been regarded as a risk factor for more severe RSV. Of note, while bronchiolitis and pneumonia are both serious, as a proportion of cases, death from RSV is relatively rare, but significantly greater than influenza in children, which is a leading cause of death for children.

A major factor in determining the clinical outcome is the titer of neutralizing maternal antibodies that are transferred across the placenta, with higher titers showing a clear relationship with less severe disease. Unfortunately, maternal antibodies are a form of passive immunity and are unlikely to be present at meaningful levels past the few months of life. Furthermore, absent an effective vaccine, the only way to boost these antibody titers would be through infection by RSV, and for the greatest boosting effect, this would have to occur relatively late in the course of the pregnancy, which is certainly not ideal. While true that there is no known adverse effect of maternal infection on the fetus, the virus is not known to cross the placenta, RSV is usually a mild illness in adults, including the pregnant, this is not universally the case.

A commonly cited reason for why very young children develop such severe manifestations of RSV has to do with their immature airways, and specifically the inability to perform collateral ventilation. Infants’ bronchioles have a diameter that is roughly half that of adult bronchioles, which means that through any given bronchiole, there is 16-fold less air flow compared with that of the adult, meaning that even small obstructions can have very profound effects and bronchiolitis is especially serious and far more likely. To compensate for this, structures called canals (channels) of Lambert, channels of Martin, and pores of Kohn exist to allow for airflow between adjacent alveoli, so that if a bronchiole becomes obstructed (e.g. from bronchiolitis), the alveolus can still perform gas exchange by using air communicated from a different bronchiole that passes to the adjacent alveolus. The problem, however, is that these structures are not well developed until about age 4 in children (however, some evidence argues that the Pores of Kohn are actually an artefact of tissue fixation and not a real anatomical structure).
Adults who develop RSV generally have a mild upper respiratory tract infection and the illness was generally milder than influenza, however, it lasted longer (9.5 days vs. 6.8 days). Influenza was also more likely to cause missed work. Despite this, in the aforementioned study, about one-fifth of previously healthy adults were found to have some form of lower respiratory tract illness.
RSV can be a major illness among older adults, with a disease burden and severity comparable to that of influenza among high-risk older adults, and in some cases even slightly higher. Older adults are generally considered to be at higher risk because they are more likely to have many of the comorbidities that make severe RSV more likely to occur, and because of immunosenescence, the set of changes to normal immunological functions that occur with advanced age. RSV in older adults is not as well understood as in children because it is under-researched and does not have distinctive clinical presentations like bronchiolitis, and thus requires specific diagnostic testing to identify (which often is not done because it would not change the management of the condition).
RSV does have some important potential health consequences outside the respiratory tract. For example, it is the most common viral cause of otitis media (middle ear infections). When RSV is particularly severe (i.e. enough to cause critical illness- not self-limiting upper respiratory tract disease), it has been observed causing a sepsis syndrome in some children, myocardial injury, neurological disorders (seizures, hyponatremia; in general the virus is not believed to infect the central nervous system although there is a case report of it being found in the CSF of an infected infant), among other problems. Additionally, RSV may be complicated by infections with other viruses and bacteria. In adults, the major bacterial superinfections are from Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, Pseudomonas aeruginosa, and Moraxella catarrhalis.
One of the longstanding controversies regarding RSV is its role in the development of asthma. RSV has long had a strong association with the development of asthma, and there is some evidence that this could be causal. The causal model argues that (in particular, lower respiratory tract) disease with RSV triggers fundamental changes to the structure and function of the airway that raises the risk of recurrent wheezing. An alternative possibility is that some children already had the predisposition to developing reactive airway disease but RSV is an effective trigger for the condition to manifest. Evidence for the causal relationship is mixed: in principle, the prevention of severe RSV would be expected to reduce the incidence of reactive airway disease, and while some evidence does support that this is the case, other data argues against it. A non-causal model on the other hand argues that the risk factors that predispose to asthma also predispose to severe RSV manifestations, and so the apparently more common cases of RSV bronchiolitis seen with children who go on to develop asthma is the result of confounding. It is generally accepted that the risk factors that predispose to asthma (atopy) also predispose to severe RSV (although they don’t appear to be the strongest risk factors for severe RSV), but this does not rule out the possibility of RSV acting as a trigger for the development of asthma in certain cases. It is also certainly possible that the relationship is bidirectional: both lower respiratory tract RSV and atopy have a common genetic cause and RSV causes atopy to progress into reactive airway disease. It’s also worth noting that RSV is not the only virus for which we wonder about the existence of this relationship. Whatever the truth of it is, RSV is clearly a major public health problem that is worth bringing under control to the extent that is possible, and an effective vaccine should be the cornerstone of our strategy.

History: The Lot 100 Catastrophe

It did not take long following the discovery of RSV for it to become apparent that RSV was a major cause of respiratory disease in young children, and so there was immediately an effort to create an effective RSV vaccine. In the not-too-distant past, the techniques available to vaccinology were more substantially more limited. Basically, there were 2 main options for making a vaccine: inactivate the pathogen (make it incapable of replicating and therefore causing disease, usually by treating it with an agent like formaldehyde to “kill” it) or make a live attenuated strain of the pathogen (culture the pathogen for many generations without the selection pressure of an immune system so that it progressively lost its virulence). If the pathogen caused disease through a toxin (like pertussis, diphtheria, or tetanus), you could also purify out the toxin and administer it in an inactivated form (called a toxoid) with an adjuvant, but this doesn’t apply to RSV. For RSV, in 1966, the first attempt was made with Lot 100, a formalin-inactivated (FI) vaccine adjuvanted with alum. It was administered by intramuscular injection as a 3-dose series with a 1 month interval between the first and second doses and then the third dose given 3 months later. It went basically as poorly as it is possible for a vaccine to go. Not only did the vaccine fail to provide protection, but the children who received it had worse outcomes: 20 of the 30 vaccine recipients developed RSV, 80% of whom had to be hospitalized, and 2 of whom died (and on autopsy, RSV was readily recovered from their lungs). In contrast, the control group, which received either a parainfluenza type 1 vaccine or a trivalent parainfluenza vaccine, the hospitalization rate was 5% and 0% respectively, and there were no reported deaths (but risk of infection for the type 1 parainfluenza vaccine group was similar). The hospitalizations in the vaccine group were also longer, at a mean of about 10.5 days compared with 6.7 in age-matched RSV infections with pneumonia or bronchiolitis. Further investigation also showed that the children who had received the vaccine at the earliest time points, between 2 and 7 months of age, tended to develop the most severe disease, hinting that the state of not having prior RSV infections was a risk factor for the development of this enhanced RSV disease (which would be confirmed by subsequent study). The Lot 100 catastrophe greatly stalled efforts for an RSV vaccine, and it was at that point decided that the only way to make a safe RSV vaccine would be through a live attenuated strain. Subsequent research would challenge that assumption.
This phenomenon, wherein the vaccine induced more severe disease, is known as vaccine-associated enhanced respiratory disease (VAERD) but it may also appear in the literature simply as enhanced respiratory disease (ERD), vaccine-enhanced disease (VED), or vaccine-associated enhanced disease (VAED).
Virological and immunological features of RSV relevant to vaccine development
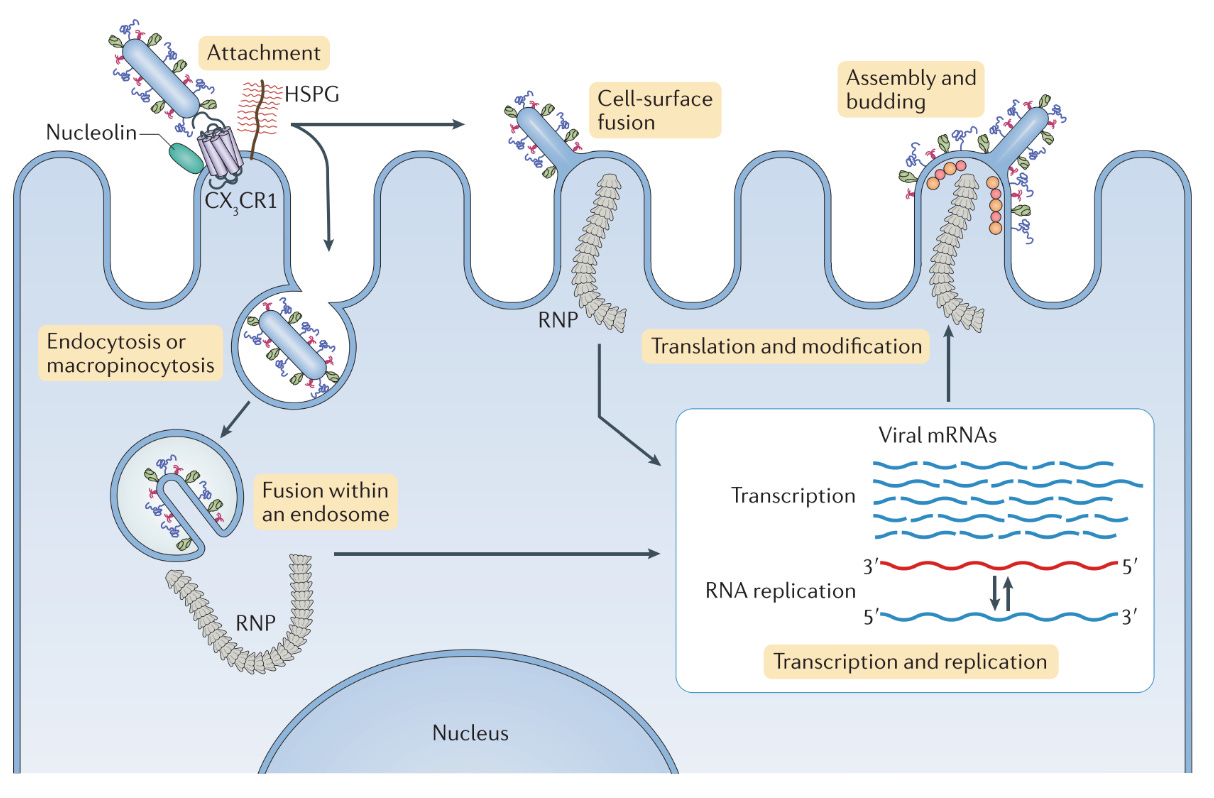
To understand what happened with RSV vaccines, it’s necessary to understand a little bit about the virology of RSV and how it causes disease and infection. This begins with cell entry, reviewed in excellent detail here. RSV is an enveloped virus in the pneumovirus family (it has been reclassified a lot over its history- older references may describe it as a paramyxovirus in the pneumovirus genus but then pneumovirus was made into a family for reasons that I am sure are valid and only make sense to the ICTV) with a negative-sense, unsegmented, single stranded RNA genome which enters cells through the use of its F (fusion), G (attachment) and SH (small hydrophobic) proteins. Of these, F and G are both potential targets for neutralizing antibodies. SH is not, but is abundantly expressed on virally infected cells, and so can be targeted by antibody-dependent cellular cytotoxicity or antibody-dependent complement activation to kill virally infected cells (and thereby arrest their further production of viral particles). The actual function of SH is not entirely clear but it is known to act as an ion channel and appears to delay apoptosis in infected cells (so that they may continue to generate infectious viral particles); deletion of the SH protein appears to reduce the ability of the virus to replicate in the lower respiratory tract of chimpanzees. The mature F protein comprises two distinct peptide chains (cleaved by furin or furin-like proteases), F1 and F2, linked together by disulfide bridges and catalyzes membrane fusion between RSV and the target host cell. RSV’s entry into host cells occurs in discrete steps through one of two major pathways. In general, the endosomal pathway is believed to be the major one:
RSV’s G protein mediates attachment via binding of host glycosaminoglycans (GAGs) found on the surface of the transmembrane proteins predominantly through its heparin-binding domain. CXC3R1 (fractalkine receptor) has also been noted to act as a possible receptor for RSV’s G protein; deficiency of CXC3R1 in mice appears to reduce the infectivity of RSV.
RSV isolates have been created lacking G and SH, however, and demonstrated that F protein is sufficient for infection as these virions still infect cells (although this is less efficient). Furthermore, treatment of heparinases does not prevent infection by these isolates, suggesting a non-GAG-mediated entry pathway; insulin-like growth factor 1 receptor (IGF1R) is a known receptor for the virus that binds F and nucleolin is a coreceptor, but there are likely others; it is a known coreceptor for the virus. In wild type RSV, because of the stereoelectronics of cell entry, RSV is believed to enter primarily with the aid of G-dependent pathways in accordance with the provocateur model of Paramyxovirus entry.
Upon stable attachment to the apical surface of the respiratory epithelial cell (the major cell type infected by RSV), the cell initiates macropinocytosis and the RSV virion enters a macropinosome.
Within the macropinosome, furin or furin-like proteases initiate a second cleavage of the F protein (the first one occurs in the normal processing of the protein), which renders the particle infections by triggering RSV F to convert to its postfusion conformation and triggering membrane fusion. This releases the RSV genome as a ribonucleoprotein complex and the infectious cycle of the virus can begin. This process takes an average of 50 minutes in vitro.
The alternative to the endosomal pathway argues that RSV enters the cell directly at the surface of the membrane via cholesterol-rich microdomains. This pathway proceeds as follows:
RSV’s G protein tethers it to the cell via CXC3R1, TLR4, or GAGs, as above, on a cholesterol-rich microdomain.
RSV F subsequently binds IGF1R, which uses signaling via PKCζ to recruit nucleolin to the cell surface to bind to RSV-F
RSV F initiates fusion at the plasma membrane and releases its genome directly into the cell.
Once inside the cell, RSV replicates entirely in the cytosol (as shown by its ability to replicate in cells that have had their nuclei removed). Adjacent RSV-infected cells may be able to fuse their membranes with one another to form syncytia; the virus does not kill infected cells efficiently. A detailed discussion of the replication of RSV once it is within the cells is not included here for brevity’s sake and because it is not directly relevant for the production of effective vaccines. Autopsy studies of fatal and non-fatal RSV infection suggest that RSV in general does not exit the respiratory tract and infection is limited to the ciliated cells of the apical epithelium (with the rare exceptions discussed earlier). The molecular mechanisms underlying the lack of susceptibility of basal cells to infection are not well understood (although the virus is known to preferentially bud out from the apical surface, which has been proposed as a mechanism by which it evades systemic immune responses), but are likely important for ensuring the infection remains confined to the respiratory tract; however, in vitro studies do demonstrate that the virus is capable of infecting the basal cells under some conditions.
RSV is an exceptionally skilled modulator of the immune response. Many respiratory viruses cause reinfections throughout the lifespan through progressive antigenic drift away from the host immune system’s coverage, but generate fairly robust immunity against the same strain of virus (such as influenza or rhinoviruses). RSV, in contrast, shows far less antigenic diversity than either of these viruses (although there is meaningful variation within the virus itself, and indeed, this is believed to have resulted in the failure of the monoclonal antibody therapy suptavumab against RSV B) and reinfects throughout the lifespan with the same strain. In particular, unlike the aforementioned viruses, the epitopes that are responsible for the most potent neutralization are well conserved despite the genetic variation across RSV isolates. Consistent, durable protection against RSV infection is never achieved- but reinfections are consistently milder than primary infections with the exception of the elderly and immunocompromised who may have severe disease despite the existence of immunological memory. Still, even in the case of the elderly, a strong association exists between the level of neutralizing antibodies against RSV and the severity of illness they experience upon infection.
Like most RNA viruses, among the first things RSV does upon entering the cell is suppress recognition of its genome by host pattern recognition receptors (RIG-I, MDA5) by disrupting signaling to MAVS that would go on to initiate an antiviral interferon response which would eventually result in complete arrest of translation and thus inability to make viral proteins for replication. RSV is also known to bind TLR4 via its F protein, but the significance of this is incompletely understood. Some work have suggested that TLR4 signaling initiated by RSV F predisposes to Th2 immune responses; overexuberant Th2 responses are associated with failure to clear the virus and are implicated in immunopathology. RSV NS1 and NS2 both disrupt both type 1 interferon and type III interferon signaling (which result in muted T cell and antibody responses); NS2 also appears to contribute to shedding of apical cells to induce obstruction of the airway.
RSV’s G protein is secreted by infected cells and can function as a “sponge” for antibodies which would be neutralizing by binding them so that they cannot bind G on RSV virions (which would interfere with infection). G is capable of binding to CX3CR1, a chemokine receptor found on multiple lineages of cytotoxic cells, and appears to be able to initiate signaling via the protein as a mimic of fractalkine, the native chemokine, recruiting these cells to the site of infection. CX3CR1 is additionally expressed on regulatory B cells, and RSV is able to infect these (aided by the tethering of its G protein) to produce more severe infections by triggering production of anti-inflammatory cytokines that inhibit virological control. RSV’s N protein also appears to play an important role in immunomodulation by disrupting the immune synapse to interfere with activation of T cells. IFN-γ is secreted by NK cells and T cells which can be recruited by the action of CX3CR1 to help control the infection; however this also results in a reduced antibody response. RSV also appears to use its G protein to suppress effective antigen presentation by altering dendritic cell phenotype towards a state of reduced activation, further limiting the induction of effective RSV-specific immunity. Furthermore, vaccinia vectored vaccines with RSV G appear to induce Th2 responses whereas those which express RSV F give Th1 responses, suggesting that the G protein inherently biases away from canonically antiviral immune responses (although in both cases, inflammatory infiltrates occurred in the lungs).

It has been suggested that among the reasons that RSV affects children so severely is not just because of a lack of RSV-specific memory to protect them (beyond what may be passed down by maternal antibodies) but rather because of a lack of maturity in their innate immune system. Specifically, it has been noted that type III IFN production may be impaired in the very young, and a protective correlation exists between positive patient outcomes and the level of type III interferons generated in response to infection. Further support for this comes from the finding that bronchoalveolar lavage fluid from infants with RSV bronchiolitis is hypercellular. Infants younger than 3 months of age produce low levels of IFNα on RSV infection.

Successful clearance of the virus in infants is shown to be temporally correlated with the appearance of neutralizing IgA antibodies in their nasal secretions. While not definitively established, a serum neutralizing antibody titer of 1:200 as measured by a 60% complement-enhanced PRNT assay is shown to be protective from severe lower respiratory tract infections. Additionally, low nasal IgA (<1:60) has been established as a risk factor for symptomatic RSV infection. In accordance with the established importance of neutralizing antibodies, several monoclonal antibody therapies exist for high risk infants (i.e. premature and born during RSV season) as passive immunization; Palivizumab is the most widely available, and targets the 101F epitope located at antigenic site A (site II) which is conserved in both pre- and post-fusion conformations. The most potent neutralizing sites however lie in prefusion F exclusive sites: Ø, III, and V, which have potencies 10 to 100 times greater than palivizumab. Motavizumab is a monoclonal antibody based on palivizumab which has undergone affinity maturation to achieve greater potency but is not used clinically. More recently, nirsevimab (which binds site Ø) has completed clinical trials for the prevention of lower respiratory tract disease by RSV in healthy and late pre-term infants and demonstrated an efficacy of 74.5% (95% confidence interval [CI], 49.6 to 87.1; P<0.001); it has been engineered to have an extended half-life and thus appears to need only a single dose administered before the beginning of RSV season for effectiveness (unlike palivizumab which has to be re-administered monthly). Interestingly, neutralization may not be the primary mechanism of action for at least some of these antibodies, as a study of palivizumab glycoforms found that a plant-synthesized form of the antibody had significantly superior control of viral load in vivo and greatly enhanced binding to FcγRIII and FcRγII, suggesting that increased neutralization potency may give diminishing returns at the therapeutic level if antiviral effector functions are inappropriately matched.

While neutralizing antibody against the F protein is likely protective, there appear to be many complexities to this observation. For example, as would be expected for a respiratory virus, nasal neutralizing antibody titers were more predictive of protection than serum antibody titers in healthy adults who were challenged with RSV; however, production of nasal sIgA was impaired (showing failure to induce in most altogether and even among responders showed unusually rapid decay kinetics) and in contrast the immune response to RSV was predominantly systemic despite its strict mucosal localization (whereas the opposite is seen for influenza). However, memory B cell (MBCs) levels pre-infection were not associated with protection or symptom severity and there was a striking absence of RSV-specific IgA+ MBCs. The source of IgA appeared to be predominantly from plasmablasts. Additionally, some evidence suggests that the key protection mechanisms from RSV change across the lifespan, as another human challenge study found that in older adults, sIgA titers were not associated with protection, and, indeed, their induction was significantly impaired compared with younger adults and associated with a higher viral load. This raises important questions about how to optimize protection from RSV with a vaccine strategy. Furthermore, once infection has occurred, antibodies do not appear to be capable of protecting from mild disease based on a challenge study of healthy adults.

Despite the apparent value of antibodies in protection, there is key evidence that protection from RSV disease is critically dependent on the function of CD8 T cells, as those who develop the most severe manifestations of RSV tend to be those with CD8 T cell deficiencies. The RSV matrix protein has been shown to be a key T cell target wherein activated CD8 T cells specific to the DbM187 epitope are able to lyse infected cells and control viral load without driving immunopathology. Intranasal administration of a murine cytomegalovirus vector expressing RSV matrix showed rapid control of the infection and demonstrated the importance of tissue-resident memory T cells in protection; critically, these responses rely on type I IFN signaling, which RSV antagonizes, as discussed above. CD8 T cells also skew the immune response away from Th2 biases which impede efficient viral clearance. Paradoxically, certain CD8 T cell responses appear to be associated with significantly worsened disease: Tc2 cells which secrete IL-4 are enriched in the airways of patients with severe RSV, whereas those who experience moderate disease have enriched Tc1, Tc17, and Th17 responses. This is also supported by the finding that monocyte-derived IL-12 appears to be an important determinant of overall clinical outcome with low levels associating with longer durations requiring ventilator use. CD8 T cell responses to RSV also decline precipitously with age. Additionally, the presence of neutralizing antibodies has been shown in mice to prevent CD8 T cell immunopathology seemingly through restriction of IFNγ production and reduced activation of memory CD8 T cells. Additionally, enriched Th9 and Th22 cell responses have been found in those who exhibit severe bronchiolitis, with CD8 cells that produced IL-9 showing a clear association with the duration of hospitalization.

Broadly, successful control of RSV infection seemingly relies on an early Th1/Th17 predominant immune response that includes tissue-resident CD8 T cells and high neutralizing antibody levels in the upper respiratory tract, with appropriate regulation to minimize immunopathology. In contrast, several cell types are associated with more severe disease and they are more likely to occur in neonates (who have a tendency towards Th2 induction). Neutrophilic inflammation of the airways appears to predispose to enhanced recruitment of CD8 T cells that drive immunopathology and suppress early Th17 responses that would ordinarily control RSV infection. The role of eosinophils in disease is incompletely defined but they are known to be a feature of severe manifestations of RSV; it is possible that the higher levels of eosinophils reflect a compensatory response to control viral load after primary mechanisms have failed given the finding that eosinophils do have antiviral activity and can capture viruses, but this function is found to be defective in asthma (which as above has complex associations with RSV). Curiously, IgE appears to be induced by some RSV infections and associates with more severe disease; it remains unclear whether the IgE is induced because the children are atopic or whether RSV induces the state of atopy. Regulatory T cells have been shown to contribute to protection by limiting immunopathology caused by activated T cells, skewing the immune response away from Th2 patterns associated with failed control, and by promoting antibody production. Group 2 innate lymphoid cells (ILCs) may hold a key piece of the puzzle in RSV immunity, as they are early responders to infection, resident in the airway, and prolific sources of IL-5 and IL-13 which are principally associated with severe RSV infection; RSV infection in mice appears to activate group 2 ILCs by inducing production of thymic stromal lymphopoeitin (TSLP). In totality, this suggests that group 2 ILCs may be a key cell type in setting the tone of the immune response against RSV.
Beyond the fact of RSV’s substantial direct propensity to function as a modifier of the host immune response, there is another issue regarding RSV F in that the protein is metastable and readily transitions from its prefusion to postfusion conformation spontaneously. As discussed above, the most potent neutralizing epitopes are accessible only in the prefusion conformation, and postfusion F tends to accumulate on the surface of the virus. This makes the elicitation of high levels of neutralizing antibodies intrinsically challenging because the critical binding sites are relatively concealed from the immune system. Furthermore, it has been demonstrated that formalin-inactivated RSV F (as was used in the Lot 100 vaccine) lacks prefusion F entirely.

Enhanced Respiratory Disease (ERD)

Enhanced respiratory diseases seen following the lot 100 vaccines was not an entirely unique phenomenon, as the killed measles vaccine was rapidly withdrawn from market after it was shown to cause atypical measles, a more severe form of measles with similar findings. This signaled that there could be something intrinsic to respiratory viruses (and consequently their vaccines) that could be driving this phenomenon. Both vaccines were formalin-inactivated, and some work has suggested that this results in the addition of carbonyl groups to the proteins that induce Th2-based type III hypersensitivity responses upon challenge. Importantly, antibody-dependent enhancement and ERD are not the same phenomenon, as ERD is not dependent on the function of Fc receptors and in humans formal proof that the process is antibody mediated is lacking. Unfortunately, the precise drivers remain incompletely understood today. Autopsy evidence from the Lot 100 vaccinees who died showed massive viral antigen load suggesting that failure to control the infection was a key component to the disease. Histopathology also showed massive infiltration of neutrophils, eosinophils, and monocytes (and heightened levels of monocyte chemoattractant proteins are found in those who experience severe forms of RSV). The role of neutrophils in immunopathology in ERD and severe RSV is contested. Transcriptional profiling of the autopsy evidence supported that the enhanced disease was driven by type 2 inflammation with IL-33 and CCL5 jumping out as a particularly important hits. In addition to these, genes responsible for the downregulation of complement factors were also found to be greatly reduced (consistent with a type III hypersensitivity response as a driver of pathogenesis). Because of the apparent importance of the type III hypersensitivity response, it has been pointed out that RSV vaccines should strive to ensure that high neutralizing antibody titers with a low proportion of non-neutralizing antibodies be sought out so that complement-mediated immunopathology would not occur on challenge. Thus the very things which would make an RSV vaccine effective may be critical to ensuring it is safe.
Often in discussion of ERD, eosinophils are regarded as a central component to the phenomenon but experimental work in mice suggests that they are dispensable as immunization with FI-RSV still produces ERD in mice lacking eosinophils (it is however worth noting that there are many key differences between mouse and human eosinophils that may influence the interpretation of these results). Instead, Th2 cells themselves are proposed as the driver through production of IL-4 and IL-10, together with overproduction of TNF-α from Th1 cells. Depletion of CD4 T cells abolished the enhanced pathology seen with RSV challenge. Dissection of the T cell subsets in other studies suggest that enhanced disease is driven by a robust Th17 and Th2 response with minimal Th1 response, and administration of IL-27 supports a shift towards Th1 and Th17 predominant responses on challenge. Hypersecretion of mucous is seen with enhanced and severe RSV disease, which is likely the result of unbalanced IL-13 production. A split virion RSV vaccine formulation adjuvanted with Addavax also demonstrated that NK cells were capable of driving enhanced disease, as their depletion reduced disease severity, pro-inflammatory cytokine production, and eosinophil infiltration.
In totality, the evidence suggests that the drivers of ERD are complex and while there are general trends that indicate that it is driven in part by failure of virological control because of insufficient antiviral activity and subsequent failure of immunoregulatory mechanisms, the precise mechanisms are incompletely resolved and it will need to remain a point of special concern in the development of RSV vaccines.
Current [as of November 2022] Vaccine Approaches
The RSV vaccine pipeline is fairly extensive with many approaches having been attempted including inactivated vaccines, vectors, subunit vaccines, virus-like particle vaccines, live attenuated vaccines, and nucleic acid vaccines. A major breakthrough however occurred with the advent of structure-based vaccine design, the practice of optimizing vaccine antigens to elicit desired immune responses. As discussed above, the immune response to RSV is complex and RSV is exceptionally skilled in modulating the immune system to prevent the acquisition of what would be protective, durable protection from reinfection. This makes vaccine design inherently challenging as it is hard to know the nature of the immune response that should be targeted with confidence. Still, many hypotheses have been tested and at least some have borne fruit. Monoclonal antibodies administered systemically consistently showed high protection against severe RSV disease (which is likely the only practical goal of an RSV vaccine), and so it makes sense to focus on eliciting them neutralizing antibodies as the mechanistic correlate of protection in RSV. In this case, with RSV, the key was the ability to stabilize the F protein in its prefusion conformation so that the most potent neutralizing epitopes would be exposed to antibodies, which was afforded by immunogen engineering. It is notable that site II and IV on RSV F do also have meaningful neutralizing activity and are shared on both the pre- and post-fusion spike yet antibodies to these sites are typically of moderate potency. This suggests that pre-fusion F-specific epitopes on are key to vaccine effectiveness for any approach that would prioritize neutralizing antibodies as a means to protection (and we simply do not know enough about any other approach to try anything different assuming that we were to apply rational design principles rather than trial-and-error). The concept of pre-fusion stabilization was devised to address a problem in HIV vaccines and has subsequently been applied with great success to SARS-CoV-2 vaccines with robust evidence that it meaningfully enhances responses, as well as vaccines for other coronaviruses, parainfluenzaviruses, Nipah virus, HIV-1, and of course, RSV vaccines.
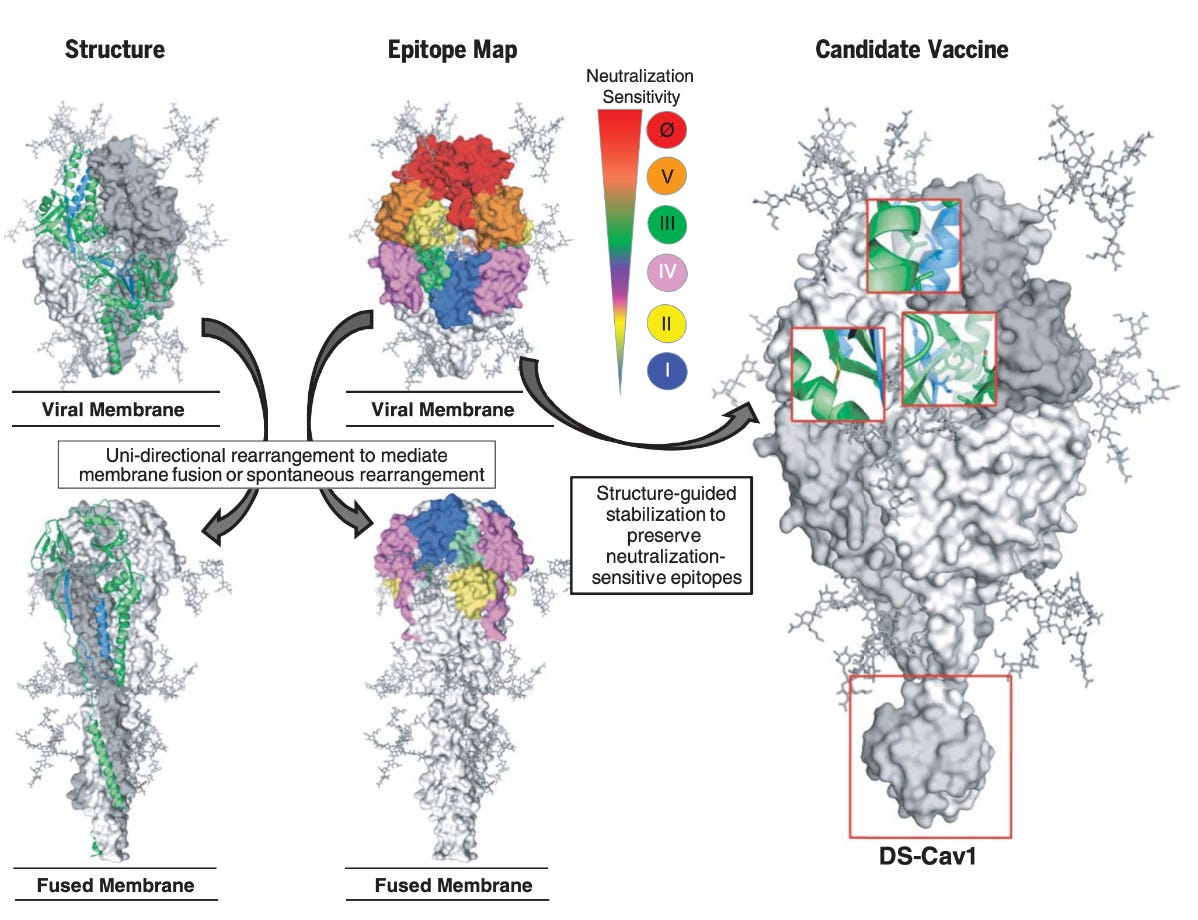
Initial efforts to engineer a prefusion stabilized RSV F antigen relied on several approaches and were spearheaded by the NIH’s Vaccine Research Center (VRC). It was critical that the antigen be highly expressed, stable against environmental conditions, and display the most potent neutralizing epitopes located in site Ø. Site Ø is present only on the prefusion conformation’s apex relative to the membrane and includes the α4–α5 loop of F1, and the F2 loop between β2 and α1. It undergoes dramatic rearrangement upon triggering α4 and the α4–α5 loop refold to form the continuous α5-helix of postfusion F. Prefusion F contains a large internal cavity and hydrophobic binding interactions within it can be used to stabilize the pre-fusion conformation to make transitioning to the post-fusion less favorable. Disulfide bonds could further stabilize it by blocking the transition, as did neutralization of an acidic patch of amino acids. This eventually gave rise to DS-Cav1, an RSV PreF which had the site Ø binding site exposed and could reliably be produced at high concentrations while also being optimized for environmental durability. DS-Cav1 subsequently underwent iterative evolution through further mutations. Another prefusion stabilized F protein was designed by targeting mutations to key refolding regions which undergo conformational changes upon transition from pre-fusion to post-fusion. Another pre-fusion F was stabilized through the introduction of prolines at specific positions, one of which (S215P) had the additional effect of greatly enhancing expression of the antigen. This exact strategy was subsequently applied to multiple vaccines for COVID-19.
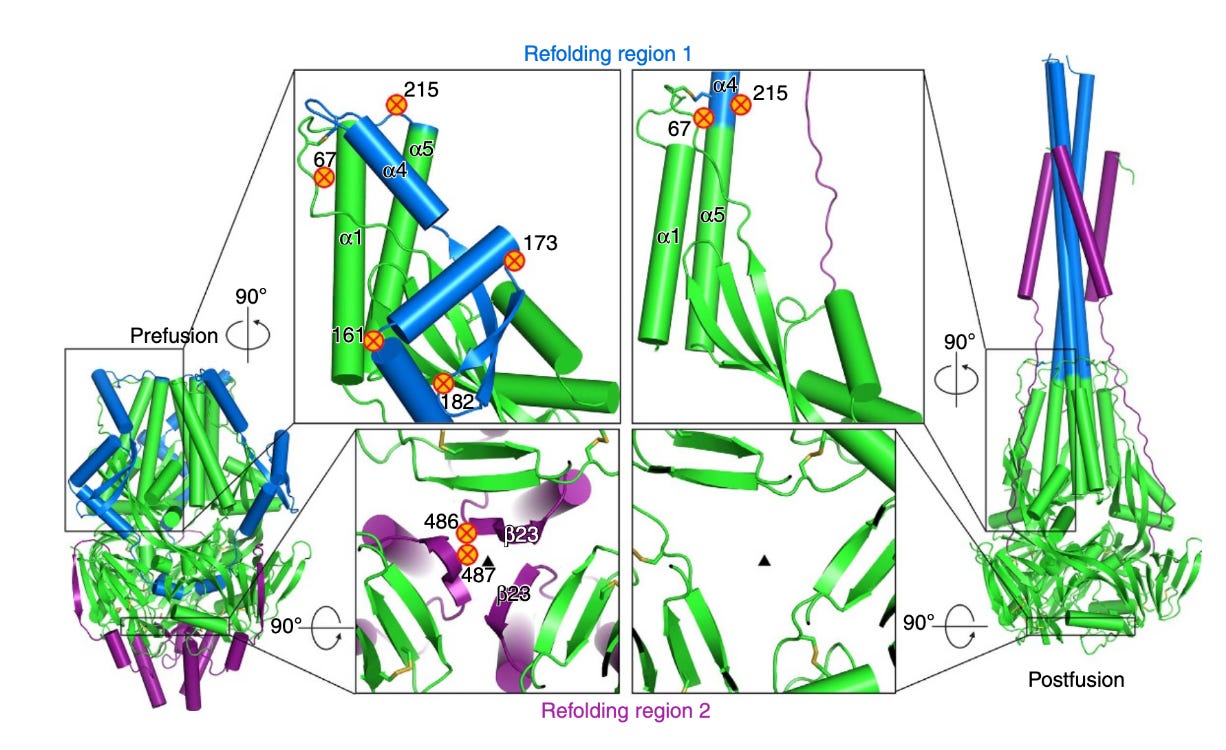
It is perhaps of special interest to discuss the two RSV vaccines which appear to have shown success in clinical trials made by Pfizer and GSK respectively. These vaccines both use prefusion stabilized RSV F (RSVPreF for Pfizer and RSVPreF3 OA for GSK); the antigens in both cases are bivalent (and of note, many vaccine candidates use RSVPreF antigen in different platforms e.g. Ad26.RSV.preF). However Pfizer’s antigen is given without adjuvant in pregnancy to protect against RSV LRTI in infants and to older adults whereas GSK’s is adjuvanted with AS01E (the same adjuvant as is used for the shingles vaccine Shingrix) for older adults only. Details on the specific constructs used to achieve prefusion stabilization are not available.
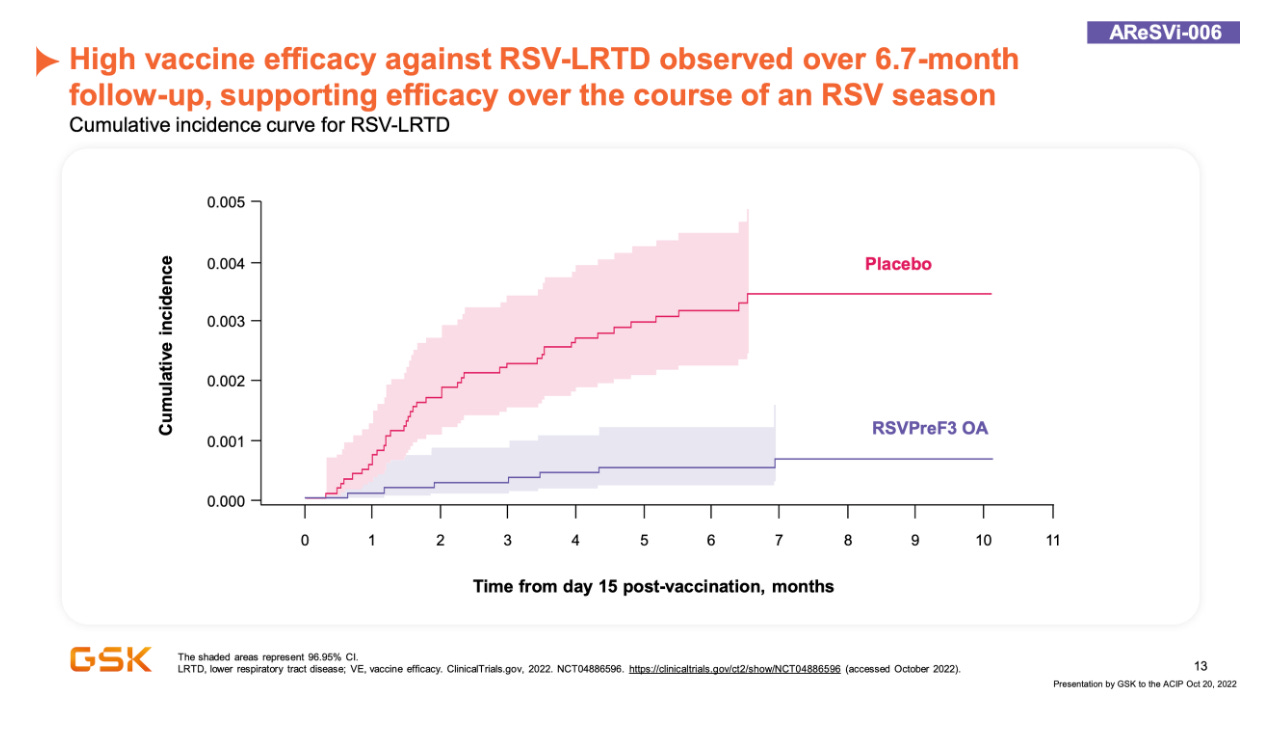
The AReSVi-006 study was a phase 3 study of the GSK’s vaccine in approximately 25,000 older adults wherein it was administered as a single 120 mcg dose to adults aged 60 or older wherein it showed 71.7% efficacy against RSV acute respiratory infection (of any severity; defined as ≥2 respiratory symptoms/signs for ≥24 hours or ≥1 respiratory symptom/sign + 1 systemic symptom/sign for ≥24 hours), 82.6% efficacy against RSV lower respiratory tract disease (defined as ≥2 lower respiratory symptoms/signs for ≥24 hours including ≥1 lower respiratory sign or ≥3 lower respiratory symptoms for ≥24 hours), and 94.1% efficacy against severe RSV lower respiratory tract disease (defined as LRTD with ≥2 LRTD signs or assessed as severe by the Investigator). All cases were confirmed by RT-PCR. As would be expected for an AS01E adjuvanted vaccine, there is substantial local reactogenicity with 60.9% of vaccinees reporting injection site pain. Additionally 33.9% of vaccinees reported fatigue within 1-2 days of vaccination. There was a slight numerical imbalance of SAEs reported across the vaccine and placebo groups, and unsurprisingly, more unsolicited AEs were observed during the first 30 days after vaccination in the vaccine group. However, no safety concerns have been identified by the Independent Data Monitoring Committee (IDMC). The vaccine has also completed a concomitant use study which suggested that it does not adversely affect immune responses to influenza vaccination and influenza vaccination also does not harm responses to the vaccine.
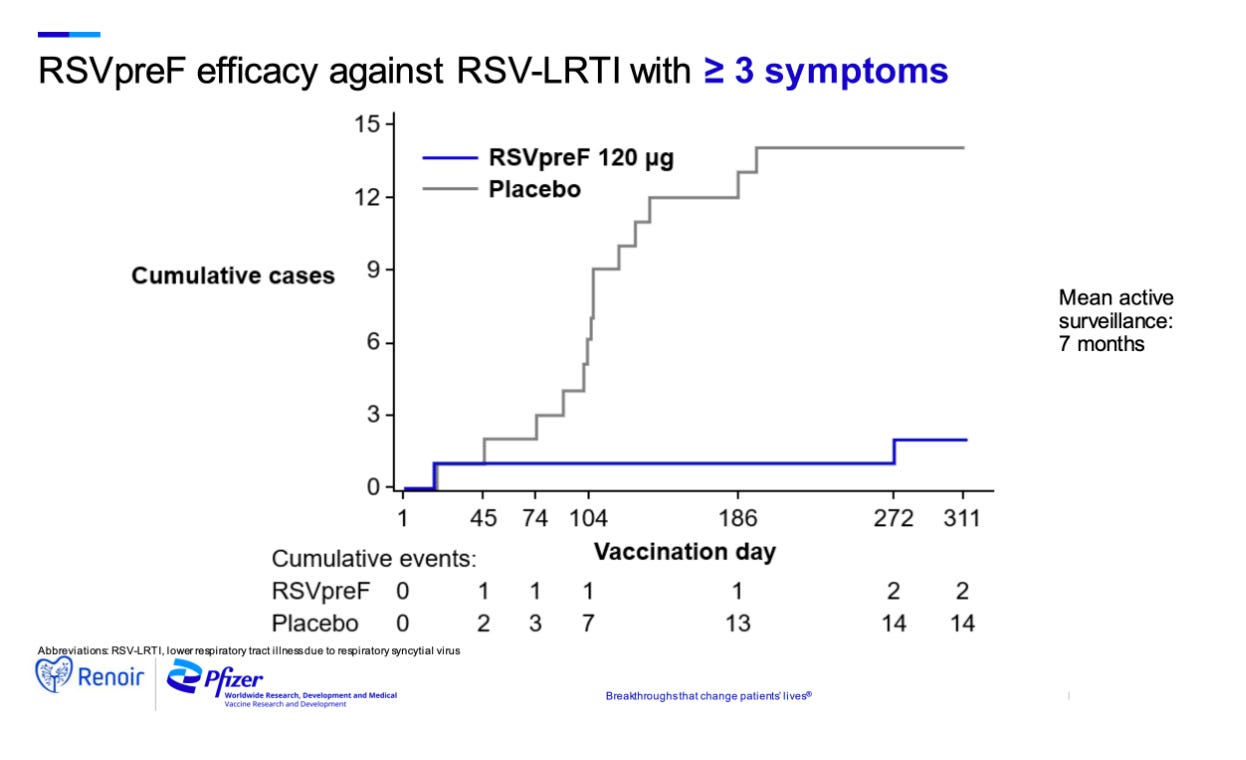
Pfizer’s vaccine was evaluated in the RENOIR study which recruited approximately 35,000 adults aged 60 or older and is administered as a single 120 mcg dose. The fact this vaccine is unadjuvanted is somewhat unusual as in general protein vaccines always require adjuvants for efficacy. I would hypothesize that this is because of RSV F’s inherent ability to bind TLR4 which may induce signaling and the presence of immunological memory against RSV F which would remove the requirement for costimulation for memory cells to induce a response, but I cannot say with certainty. As would be expected, the unadjuvanted vaccine has a milder reactogenicity profile compared with GSK’s with mainly local reactogenicity and only slight differences in the systemic reactogenicity compared with placebo. The vaccine showed an efficacy of 66.7% (28.8%, 85.8%) against RSV LRTI with at least 2 symptoms and 85.7% (32.0%, 98.7%) for RSV LRTI with at least 3 symptoms. The vaccine did have 3 SAEs which investigators assessed as being possibly related: one was a hypersensitivity response with a delayed onset, another was Miller-Fisher syndrome diagnosed retrospectively with Brighton Criteria Level 4 (i.e. very poor certainty) and one case of Guillain-Barre syndrome with an NSTEMI (Brighton Criteria Level 1 i.e. certain). The IDMC however did not feel there were safety issues and advised enrollment into the study continue. The vaccine had previously been evaluated with alum (wherein the presence of alum appeared to reduce antibody transfer to the fetus, likely as a consequence of class switching differences) and CpG DNA with alum adjuvants but these did not sufficiently boost the immune response over the antigen alone. RSVPreF has also been evaluated in concomitant use studies with influenza vaccination and no substantial difference in titer has been observed. RSVPreF has also been used in a human challenge study of healthy adults in which it demonstrated exceptionally high efficacy against symptomatic RSV infection of 86.7% (95% CI, 53.8 to 96.5) as well as a 20.5 (95% CI, 16.6 to 25.3)-fold rise in neutralizing antibody titer compared with the placebo group’s unchanged titer at 28 days following challenge (once again supporting the exceptional capacity of RSV to avoid induction of immune responses). Most recently, Pfizer announced in a press release that this vaccine showed an efficacy of 81.8% against severe medically attended lower respiratory tract illness due to RSV in infants from birth through the first 90 days of life with an efficacy of 69.4% demonstrated through the first six months of life and no safety concerns. The vaccine did not, however, meet the second primary efficacy endpoint of medically attended lower respiratory tract illness due to RSV, showing 57.1% (CI: 14.7%, 79.8%) for the first 90 days and 51.3% (CI: 29.4%, 66.8%) over the 6-month follow up period. Judging by the confidence intervals however, this most likely reflects an insufficient number of RSV cases rather than a lack of efficacy from the vaccine. Nonetheless, given the complete absence of any effective RSV vaccines, the significant effectiveness against severe disease, and the grave public health threat this disease poses to very young children this vaccine is likely worth approving, assuming that all the data prove above board upon review by FDA, CDC, and any relevant advisory committees and Pfizer has announced that it will be seeking regulatory approval.
Why is RSV back with a vengeance this season?
The short answer is: no one is certain. There are a few possibilities. One is that RSV simply became more virulent- new variants emerged with novel properties that allowed for it to cause severe disease. Though it is thought that some strains of RSV can be more virulent than others, genomic data for RSV surveillance are quite lacking and no one has (as far as I can tell) carried out the epidemiological work necessary to judge whether or not this is the case. Furthermore, it’s also possible that RSV isn’t actually any more severe and we’re just experiencing significantly higher case counts, but the same proportion of them are becoming severe disease (as RSV NET surveillance relies on hospitalized cases and we aren’t testing enough people to get a good sense of community spread). Furthermore, unfortunately there isn’t, as far as I am aware, anyone doing wastewater tracking of RSV currently, which is a shame because it both has precedent and is not dependent on case reporting. Without knowing the answers to these basic questions, our ability to explain the current RSV situation is greatly hampered. Still, beyond the direct virological explanations (which are, in my opinion, a bit lazy and improbable), there are more mundane possibilities.
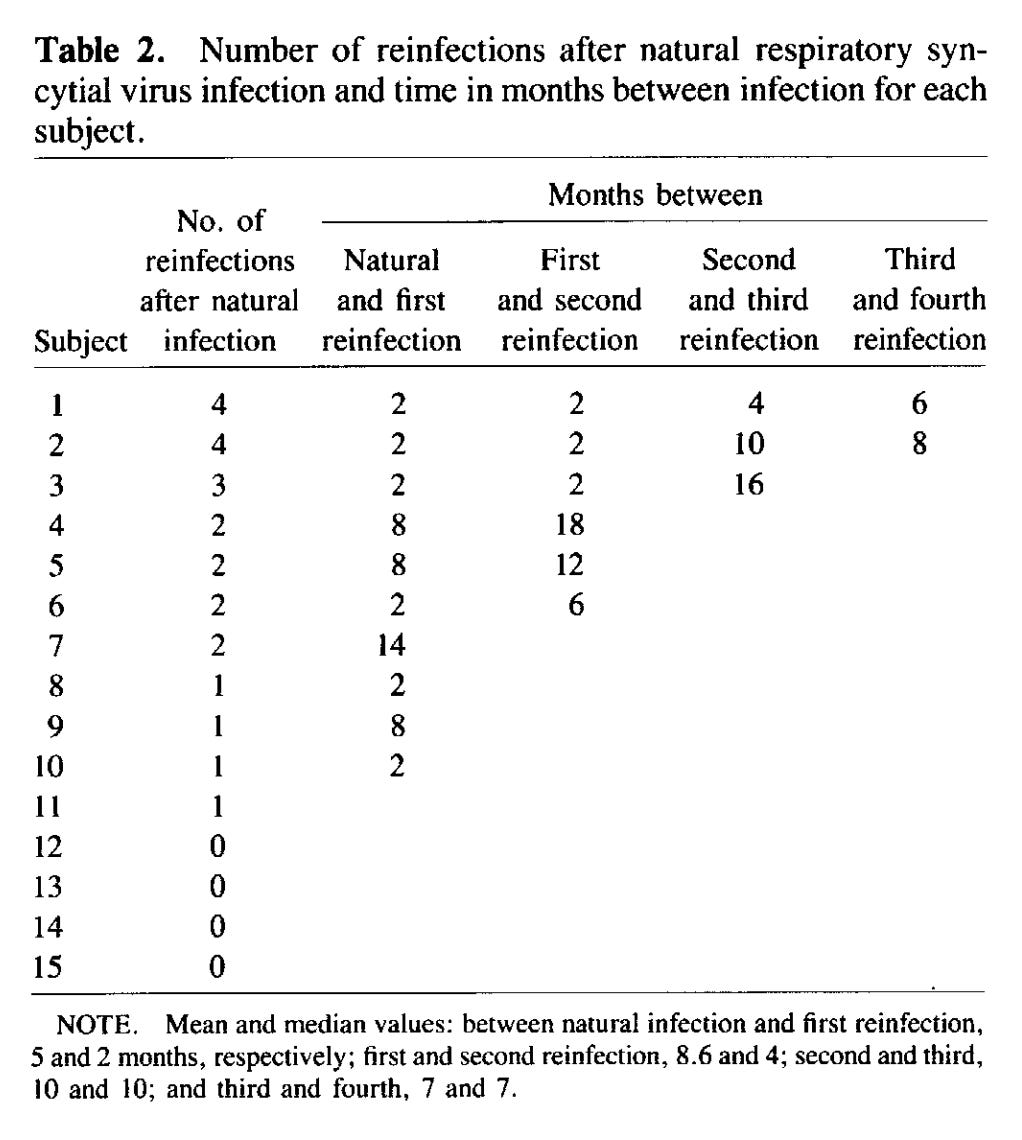
The idea that some infections require regular boosting by re-exposure to the pathogen isn’t a new one. Hope-Simpson famously proposed this as a potential issue with varicella vaccination, wherein vaccinating kids against varicella would trigger zoster outbreaks in the elderly because the elderly would not get boosted by their exposure to varicella-infected children (an issue that is easily remedied by the existence of a highly effective zoster vaccine for the elderly regardless). RSV immunity to infection is not durable. The virus is an exquisitely talented immunomodulator, and, as discussed earlier, the metastability of its fusion protein and its ability to secrete its G protein to act as a neutralizing antibody sponge greatly hinder our ability to make robust, durable responses against it, and so we are all frequently reinfected throughout our lifespan. In fact, studies of children even found that the severity of a reinfection one year after an initial infection did not seem to affect the severity of RSV, and it was not until a third reinfection that the severity of disease was found to be appreciably reduced. Experimental challenge studies reinforce this: 15 healthy adult volunteers who experienced RSV infections were infected with the A2 strain (a strain of RSV known to be less virulent and indeed used in multiple candidate live attenuated RSV vaccines) of RSV at 2, 4, 8, 14, 20, and 26 months after their initial RSV infection. At the 2 month mark nearly half of the participants had been reinfected by RSV (defined as a four-fold rise in F antibody levels) and all of these individuals shed infectious virus. At 14 months, 30% of them had been infected and 8% shed infectious virus. Fortunately, none of these experimental infections produced anything worse than a mild cold, about half were asymptomatic, and shedding was relatively rare. However a few things are important to bear in mind about these results: firstly, this is experimental infection with a virus that had undergone serial passaging in culture to become attenuated (and thus may not reflect the severity of an actual RSV infection which could be worse) and in healthy adults (who doubtlessly have an extensive history of regular re-exposures to RSV that boost their immunity, especially cell-mediated immunity, in contrast to young children who have had fewer exposures to the virus). Note that the strain of virus used for each challenge was the same one- these results are not explained by antigenic drift as they might be for influenza or SARS-CoV-2. One caveat to this data however is because the virus is attenuated, its replication may be more limited upon inoculation and thus the immune response it elicits could be more muted than what might be expected for a wild type RSV infection.
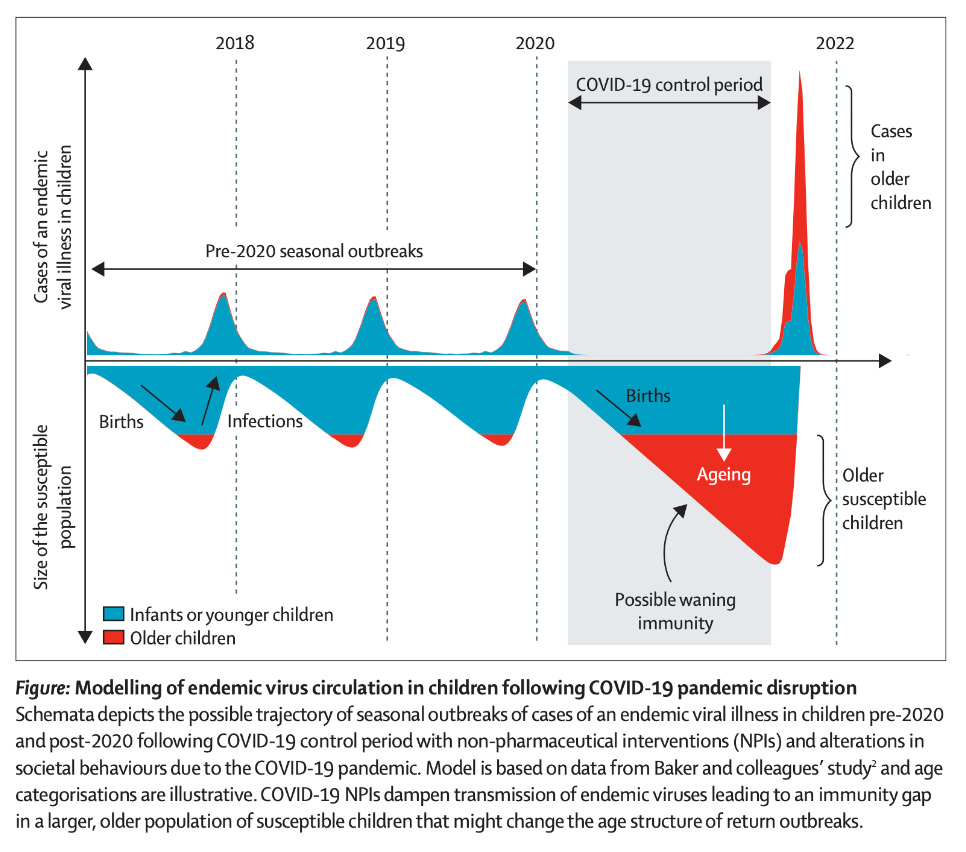
With each reinfection, our immunity to RSV is boosted for a little while and we hold on until the next season (although clearly sometimes it might not be even that long). However, this puts younger children at much greater risk for severe RSV. The gap in boosting created by an absence of RSV circulation means that mothers’ RSV antibodies that would be transferred across the placenta decay to significantly lower levels, weakening their protection. In some cases, young children age out of the protection conferred by maternal antibodies altogether and now their immune system has to fend entirely for itself without the aid of these antibodies, which helps to explain why older children than those who are normally known to be at high risk for severe RSV disease are now developing it. In fact, this was predicted before this rise even happened by Messacar and colleagues, who presciently noted:
Health systems must prepare for the potential of larger-than-typical non-seasonal outbreaks in the future due to larger susceptible populations of children being simultaneously exposed to endemic viruses. The size and timing of impending outbreaks of specific pathogens are difficult to predict because they depend on many dynamic factors, including the strength and duration of NPIs and pathogen seasonality and transmissibility.

The US’s RSV hospitalization trends on RSV NET show that the 2021-2022 season had an early and sustained peak. I therefore would not rule out that a lack of boosting to RSV from this gap, whatever the cause of that relative lack is, could be responsible for the current phenomenon we are seeing with RSV. This actually has precedent in infectious diseases. For example, hygiene initially made polio worse. Polio is typically spread mainly by the fecal-oral route, and before standards improving the cleanliness of water, nearly every child was exposed to the virus in infancy- a period in which they had maternal antibodies (both breastmilk sIgA and transplacental IgG in their circulation) to protect them from the devastating consequences of paralysis. Upon the improvement in hygiene, polio vanished from the water, and so people were exposed later in life- now without the protection of maternal antibodies- and so paralytic cases of polio became more common. This was also seen with rubella: early rubella elimination strategies focused on vaccinating infants because they are the natural reservoir for the virus (even though the reason for the vaccine is infection of the fetus in utero). As uptake of the vaccine rose, there was indeed decreased circulation of rubella- but those rare rubella cases which did occur happened to pregnant individuals with lower antibody titers to the virus and thus more limited ability to protect their fetuses.
Nonetheless, it is worth noting that RSV during the 2020-2021 season was virtually nonexistent, and given that there was no specific pharmaceutical intervention for RSV (i.e. a vaccine), the only reasonable alternative explanation I can think of to explain the absence is the success of nonpharmaceutical interventions. Given that success, and given the current situation with respiratory viruses in the US, it is of the utmost importance that we institute these measures in full force, at the very least until the beginning of 2023, at which point it is likely that we will finally have a safe, effective RSV vaccine for the next season.

I do want to address one additional item on this point, though. Some have used this situation to argue that the current situation proves that our mitigation measures against COVID-19 were a mistake. This is nonsense. Firstly, given what happened to respiratory viruses across the board, they clearly worked (I addressed this before in this post). Furthermore in parallel comparisons of nations with similar characteristics that took opposite approaches to NPIs, it’s clear that those adopting NPIs fared significantly better. The suggestion that we shouldn’t have done this is a bit like looking at a rise in paralytic polio cases upon the improvement in hygiene and calling for us to put excrement back into the water.
One other idea proposed is that this is the result of immune dysregulation caused by COVID-19. Outside of critical illness (in which case, the relevant patients are generally not going to be community-dwelling children- the demographic being hit hardest by the current RSV wave), the evidence for any substantial immunological damage caused by COVID-19 is extremely poor. COVID-19 has been truly horrible in myriad ways, but fortunately damage to the immune system does not, in general, seem to be among them, and, as discussed above, it is not necessary to explain the current situation (and thus we should not jump to it). Many studies have been done to investigate SARS-CoV-2’s effects on the immune system, almost all of which rely on in vitro work and are too contrived to reliably make any conclusions about how it actually behaves in humans. I cannot exhaustively go through each publication on this issue because it would take too long but I will discuss the two that people usually resort to to support their claims that SARS-CoV-2 does directly damage the immune system:
The first one claims to show that SARS-CoV-2 is able to directly infect T cells without the use of ACE2 and this explains some of the declines in T cell count seen in infections, but there are a number of problems here, addressed in detail by Dr. Alexander Schafer. To give an abridged summary, a major issue is that their methods appear to show significant levels of T cell infection in uninfected controls, raising major questions about the veracity of these findings, and evidence of an actual infection here is quite slim. For example, T cells were identified by their expression of CD3 and stained for SARS-CoV-2 nucleocapsid- but, there exist γδ T cells expressing CD3 which are phagocytic and can take up virus from the external environment without becoming infected. Furthermore, infection has previously been shown to produce a very large γδ T cell response, consistent with this finding. In addition to these points, the decline in T cell counts seen during acute infection should not be presumed to reflect a death of the T cells- it happens in virtually every acute, non-systemic viral infection and is generally the result of T cells trafficking out of the blood and going to the site of infection and adjacent lymphoid organs. Lymphocytes are even sometimes described as a negative acute phase reactant (meaning their levels decrease during periods of acute inflammation). In severe COVID-19, there does indeed appear to be death of the T cells, but T cells are associated with virological control of the infection, which while not overtly contradicting the idea that they are a major target of infection, raises suspicion about the idea. Additionally, T cell death is most likely the result of hyperinflammation triggering activation induced cell death, as the treatment of critically ill patients with tocilizumab is associated with enhanced recovery of the T cell counts and function, as has been seen with infliximab as well. Perhaps most importantly, in the vast majority of patients, following recovery from COVID-19, T cell counts normalize to pre-infection levels except for a subset of patients with critical COVID-19 whose CD8 T cell counts remain low which appears to be related to T cell exhaustion, suggesting perhaps a subset of patients who cannot completely clear the infection (supported by the predominance of elderly individuals and male sex in this group). Furthermore, multiple studies which have appropriate controls and experimental procedures have attempted experimental infection of T cells with SARS-CoV-2 and… it doesn’t happen. In short, our current knowledge of COVID-19’s effects on T cells is not consistent with the idea that the widespread infections of SARS-CoV-2 in children produced the current picture with RSV, and furthermore these are unnecessary to explain the current picture.
The other study claims to show that SARS-CoV-2 infection results in B cell dysfunction by downregulation of CD19. CD19 is probably best known for being a B cell marker on flow cytometry and it is part of the B cell co-receptor complex with CD21 and CD81. Its function remains incompletely understood, but it is generally regarded as a protein important for enhancing signaling through the B cell receptor (the membrane-bound form of an antibody) and thus leading to things like B cell proliferation and metabolic adaptation upon becoming activated. This work is certainly interesting, and raises important questions that are worth investigating further, but I have a few major issues with it. Per the methods, blood samples were obtained 10 to 12 weeks following recovery, but there is no information given about the severity of these infections. The majority of SARS-CoV-2 infections today are mild and self-limiting thanks to widespread population immunity, and all that we know of the patients in this study is that they were recruited from a hospital (however, this says nothing about the severity of their illness as in China all patients did go to hospital for management as part of infection control measures). Additionally, no information is provided about the vaccination status of these patients, nor the actual time period when these blood draws were taken- was anyone in this study vaccinated? Furthermore, several real-world findings cast doubt on the generalizability of these findings. For example, recovered patients who are subsequently vaccinated have exceptional antibody responses to SARS-CoV-2, which is inconsistent with the finding of a major B cell dysfunction caused by the infection. It is not terribly farfetched that there might be a transient lowering of normal immunological function following the resolution of an acute infection, but at present, quality evidence to support that SARS-CoV-2 is destroying our immune systems does not exist and should not be presumed to be the case.
To pre-emptively address the straw men that will follow my arguments that SARS-CoV-2 infections do not explain this respiratory virus season: SARS-CoV-2 is a major cause of morbidity and mortality in children and they absolutely should be vaccinated against it. It is still associated with myriad medical problems (many of which we have no good way to treat currently) and creates massive strain on our medical system, so to the extent it can be avoided, it should be. Furthermore, the prospect of superinfection with SARS-CoV-2 and either RSV or influenza or the trifecta as well as any bacteria or fungi that also hitch a ride is a truly concerning prospect that should be avoided, and that risk can greatly be reduced by reducing the risk of acquiring those infections. Vaccination against both SARS-CoV-2 and influenza are the most important and valuable protective measures that must be taken for the protection of the community and for the individual.
Now what?
My earnest hope is that the RSV vaccines pass muster in regulatory review as being safe and effective and we urgently incorporate them into routine immunization for the pregnant, elderly, and young children as a priority, and by all indications, this should be the case by about early 2023. In the interim though, the US and many countries abroad face a major challenge with respiratory viruses following the relaxation of non-pharmaceutical interventions. We have long treated the deaths each season from influenza as being unavoidable, but the pandemic has shown they aren’t. With the resurgence of all of these viruses, we should make every attempt to forestall further transmission of RSV and other viruses of serious public health concern until such time as a safe, effective RSV vaccine is available for those at highest risk, and ideally beyond that. As a reminder, this does not simply mean wearing high-quality masks. In fact, given that RSV is so often a disease of very young children, I’m not sure how effective masking can be as many children are simply too young to wear any of them. Furthermore, even if they are willing, wearing a well-fitted mask for many hours might be a tall order for children, and if done improperly, its role as infection control is essentially theater. Adults on the other hand, I don’t feel have any good justification to avoid mask-wearing in high-risk settings, especially if they work in childcare. However, fomite transmission does play important roles in the transmission of RSV and many other viruses, so thorough handwashing and surface disinfection should be emphasized (noting that these are less effective for SARS-CoV-2 which is predominantly aerosol-driven). Humidification and ventilation also remain critical for the control of transmission of respiratory viruses and should continue to be emphasized. This promises to be a very trying season for respiratory viruses- we should do everything we can to try to mitigate that burden.